

The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9
- PMID: 20089923
- PMCID: PMC6633112
- DOI: 10.1523/JNEUROSCI.3711-09.2010
The type I interferon-alpha mediates a more severe neurological disease in the absence of the canonical signaling molecule interferon regulatory factor 9
Abstract
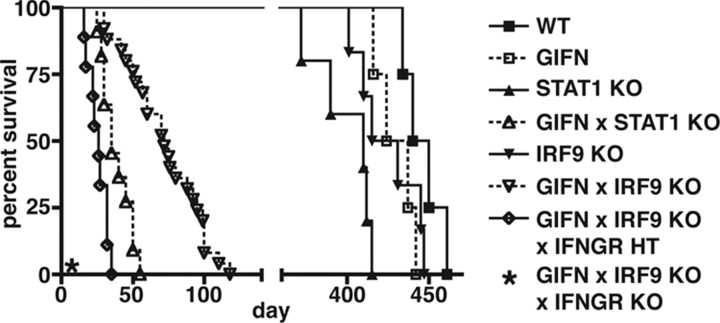
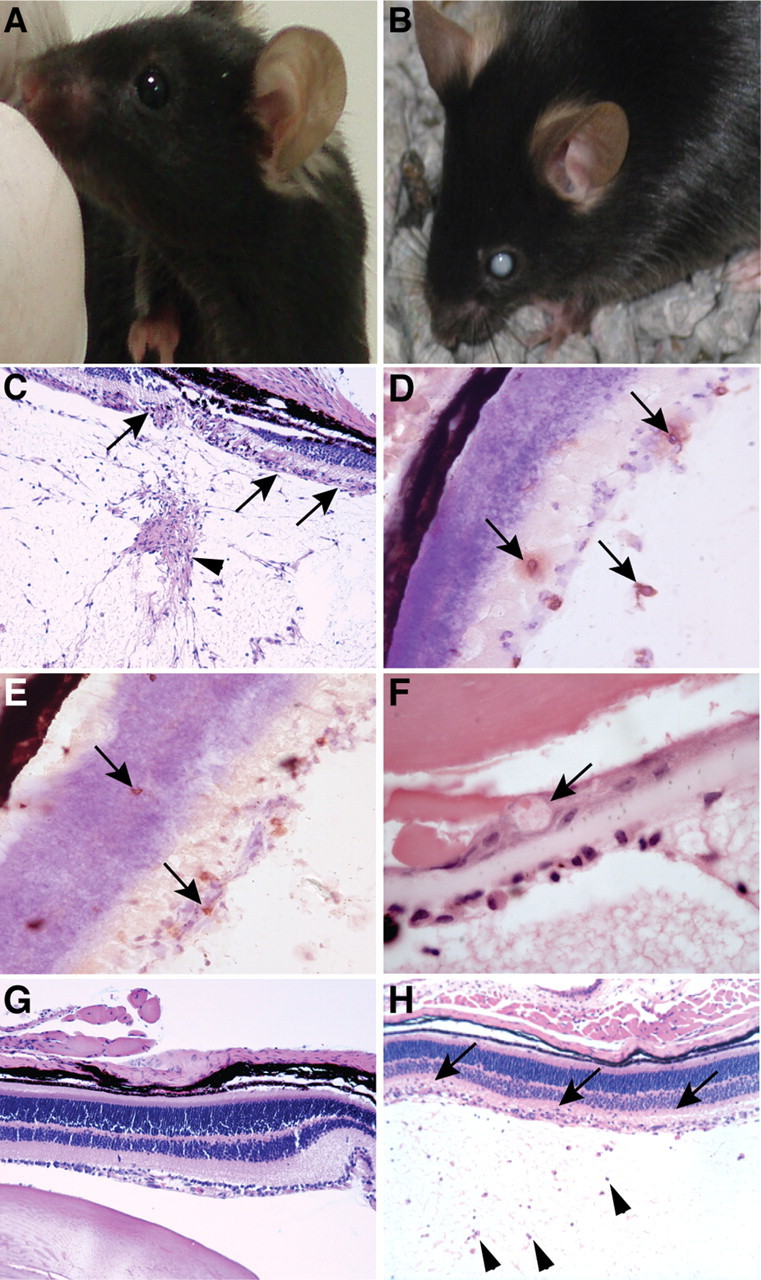
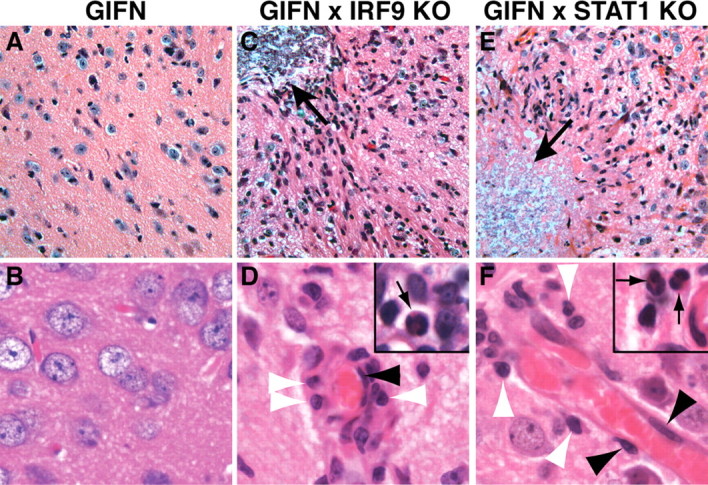
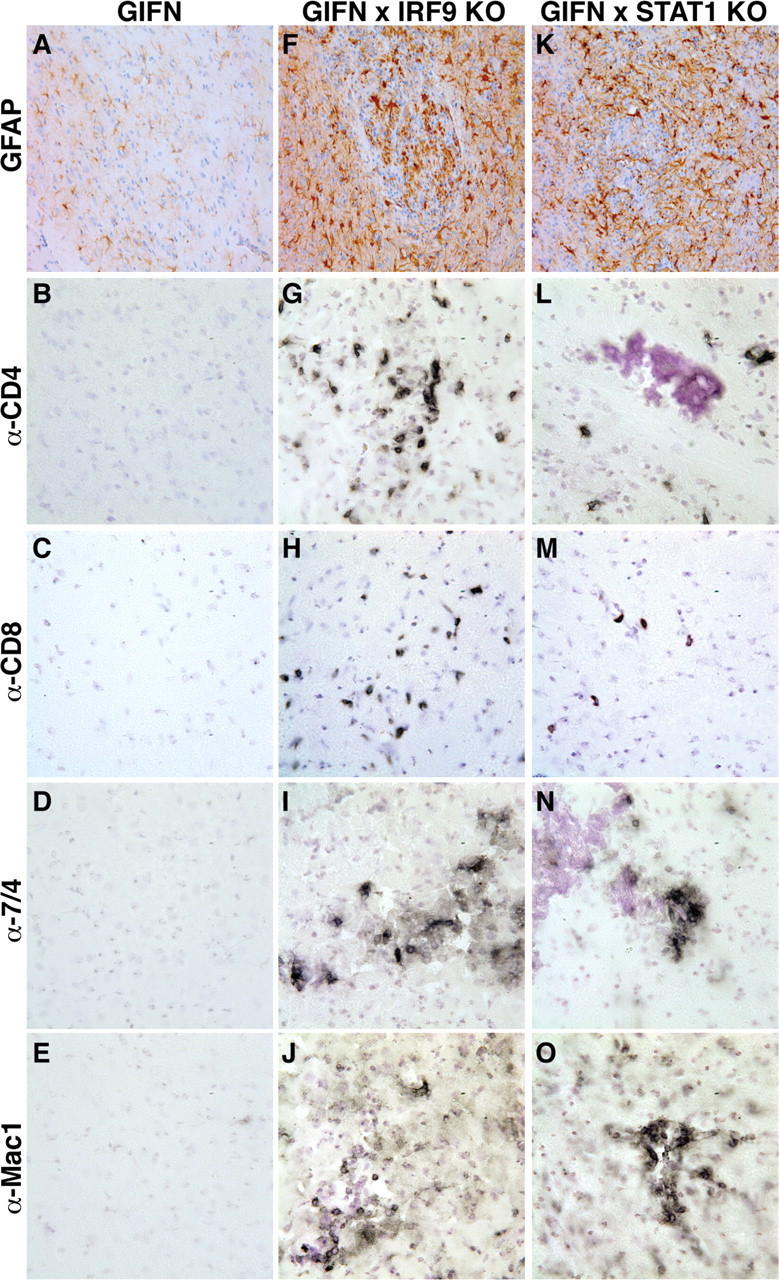
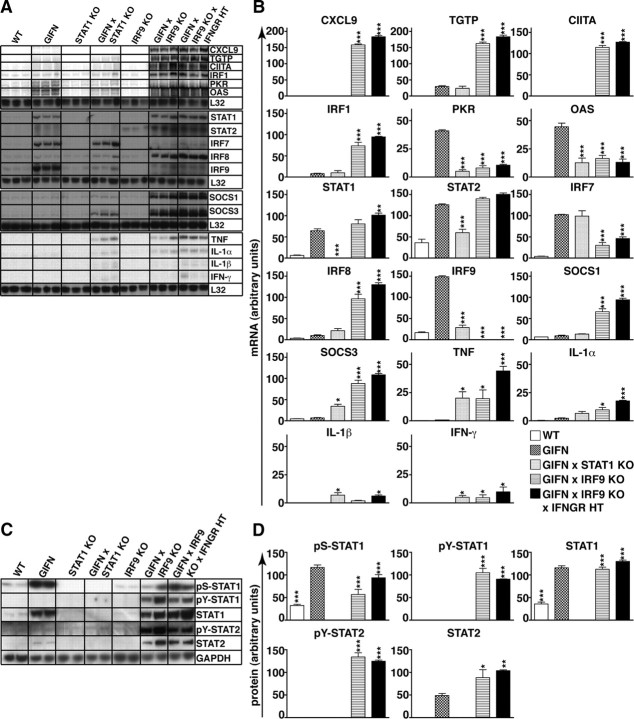
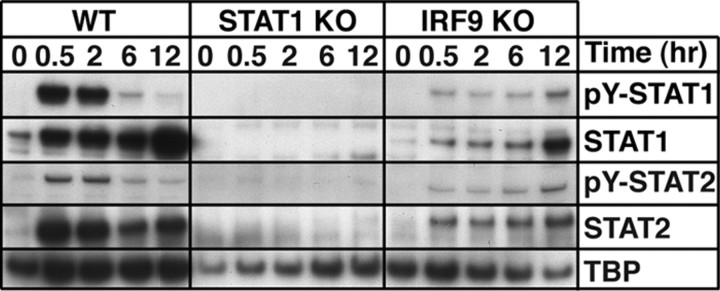
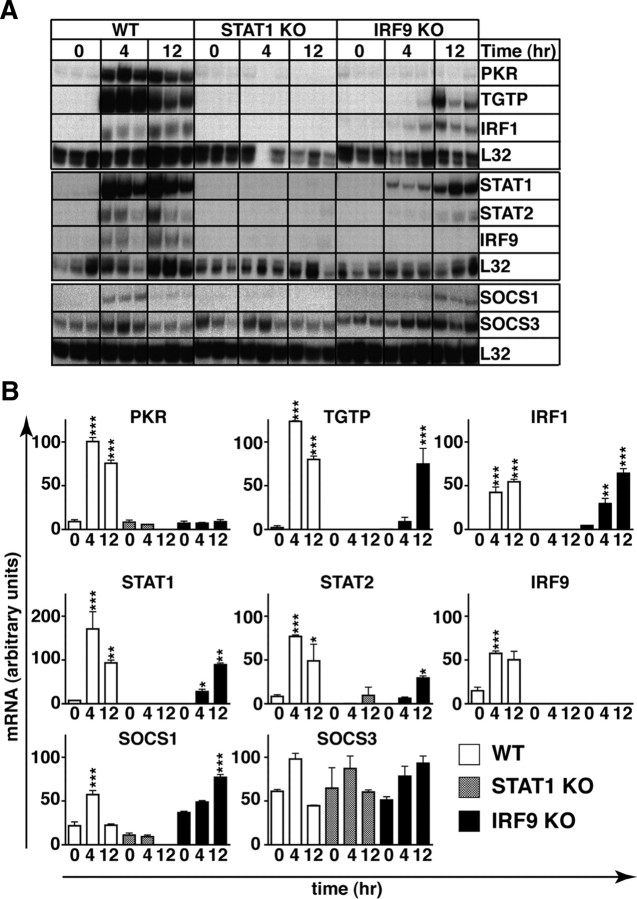
Type I interferons (IFN) are crucial in host defense but also are implicated as causative factors for neurological disease. Interferon regulatory factor (IRF9) is involved in type I IFN-regulated gene expression where it associates with STAT1:STAT2 heterodimers to form the transcriptional complex ISGF3. The role of IRF9 in cellular responses to type I IFN is poorly defined in vivo and hence was examined here. While transgenic mice (termed GIFN) with chronic production of low levels of IFN-alpha in the CNS were relatively unaffected, the same animals lacking IRF9 [GIFNxIRF9 knock-out (KO)] had cataracts, became moribund, and died prematurely. The brain of GIFNxIRF9 KO mice showed calcification with pronounced inflammation and neurodegeneration whereas inflammation and retinal degeneration affected the eyes. In addition, IFN-gamma-like gene expression in the CNS in association with IFN-gamma mRNA and increased phosphotyrosine-STAT1 suggested a role for IFN-gamma. However, GIFNxIRF9 KO mice deficient for IFN-gamma signaling developed an even more severe and accelerated disease, indicating that IFN-gamma was protective. In IRF9-deficient cultured mixed glial cells, IFN-alpha induced prolonged activation of STAT1 and STAT2 and induced the expression of IFN-gamma-like genes. We conclude that (1) type I IFN signaling and cellular responses can occur in vivo in the absence of IRF9, (2) IRF9 protects against the pathophysiological actions of type I IFN in the CNS, and (3) STAT1 and possibly STAT2 participate in alternative IRF9-independent signaling pathways activated by IFN-alpha in glial cells resulting in enhanced IFN-gamma-like responses.
Figures
Similar articles
-
Type I interferon-regulated gene expression and signaling in murine mixed glial cells lacking signal transducers and activators of transcription 1 or 2 or interferon regulatory factor 9.J Biol Chem. 2017 Apr 7;292(14):5845-5859. doi: 10.1074/jbc.M116.756510. Epub 2017 Feb 17. J Biol Chem. 2017. PMID: 28213522 Free PMC article.
-
STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1.Biochem J. 2015 Mar 15;466(3):511-24. doi: 10.1042/BJ20140644. Biochem J. 2015. PMID: 25564224 Free PMC article.
-
Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus.J Virol. 2012 Jun;86(12):6932-46. doi: 10.1128/JVI.07147-11. Epub 2012 Apr 11. J Virol. 2012. PMID: 22496215 Free PMC article.
-
The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses.Cytokine Growth Factor Rev. 2016 Jun;29:71-81. doi: 10.1016/j.cytogfr.2016.02.010. Epub 2016 Mar 18. Cytokine Growth Factor Rev. 2016. PMID: 27053489 Review.
-
A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses.Front Immunol. 2018 May 28;9:1135. doi: 10.3389/fimmu.2018.01135. eCollection 2018. Front Immunol. 2018. PMID: 29892288 Free PMC article. Review.
Cited by
-
Breaking down the cellular responses to type I interferon neurotoxicity in the brain.Front Immunol. 2023 Feb 3;14:1110593. doi: 10.3389/fimmu.2023.1110593. eCollection 2023. Front Immunol. 2023. PMID: 36817430 Free PMC article. Review.
-
The prototypical interferonopathy: Aicardi-Goutières syndrome from bedside to bench.Immunol Rev. 2024 Oct;327(1):83-99. doi: 10.1111/imr.13413. Epub 2024 Oct 29. Immunol Rev. 2024. PMID: 39473130 Review.
-
Altered increase in STAT1 expression and phosphorylation in severe COVID-19.Eur J Immunol. 2022 Jan;52(1):138-148. doi: 10.1002/eji.202149575. Epub 2021 Nov 17. Eur J Immunol. 2022. PMID: 34676541 Free PMC article. Clinical Trial.
-
Post-COVID-19 Parkinsonism and Parkinson's Disease Pathogenesis: The Exosomal Cargo Hypothesis.Int J Mol Sci. 2022 Aug 28;23(17):9739. doi: 10.3390/ijms23179739. Int J Mol Sci. 2022. PMID: 36077138 Free PMC article. Review.
-
Bioluminescent imaging reveals divergent viral pathogenesis in two strains of Stat1-deficient mice, and in αßγ interferon receptor-deficient mice.PLoS One. 2011;6(9):e24018. doi: 10.1371/journal.pone.0024018. Epub 2011 Sep 7. PLoS One. 2011. PMID: 21915277 Free PMC article.
References
-
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. - PubMed
-
- Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. - PubMed
-
- Asensio VC, Kincaid C, Campbell IL. Chemokines and the inflammatory response to viral infection in the central nervous system with a focus on lymphocytic choriomeningitis virus. J Neurovirol. 1999;5:65–75. - PubMed
-
- Asensio VC, Maier J, Milner R, Boztug K, Kincaid C, Moulard M, Phillipson C, Lindsley K, Krucker T, Fox HS, Campbell IL. Interferon-independent, human immunodeficiency virus type 1 gp120-mediated induction of CXCL10/IP-10 gene expression by astrocytes in vivo and in vitro. J Virol. 2001;75:7067–7077. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous