

Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding
- PMID: 20089953
- PMCID: PMC3156560
- DOI: 10.1056/NEJMoa0907705
Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding
Abstract
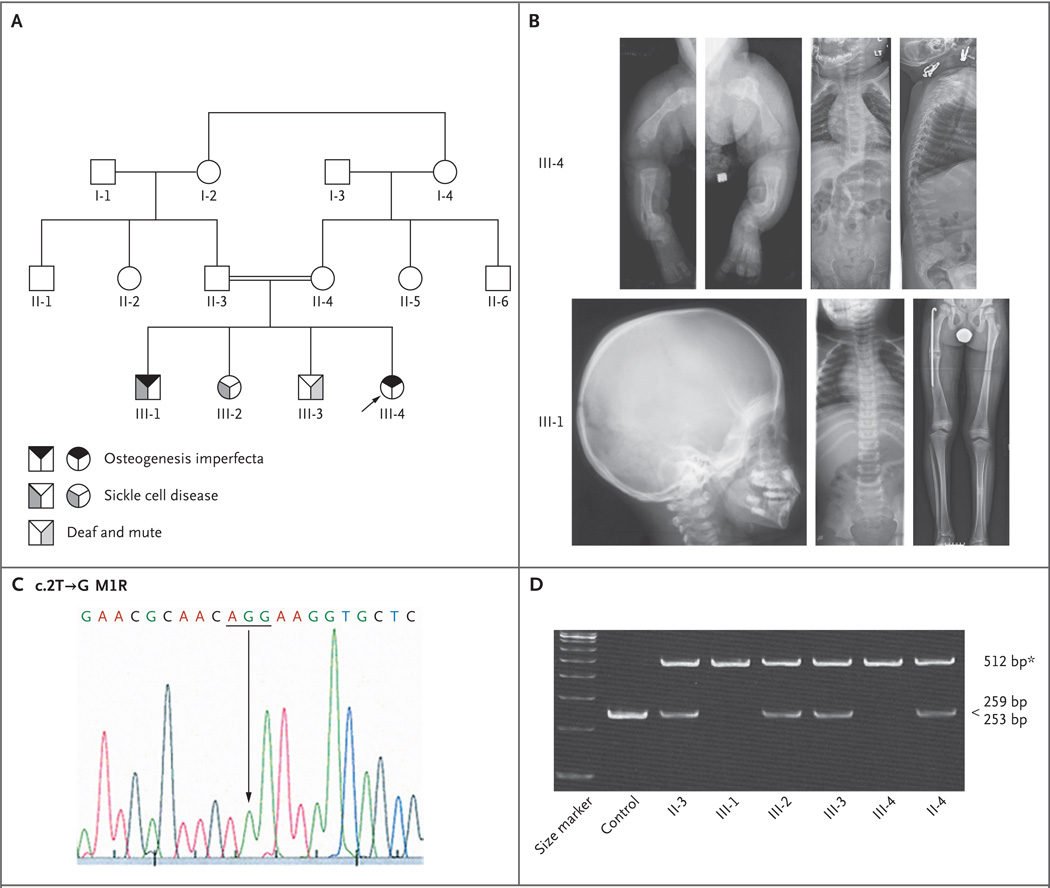
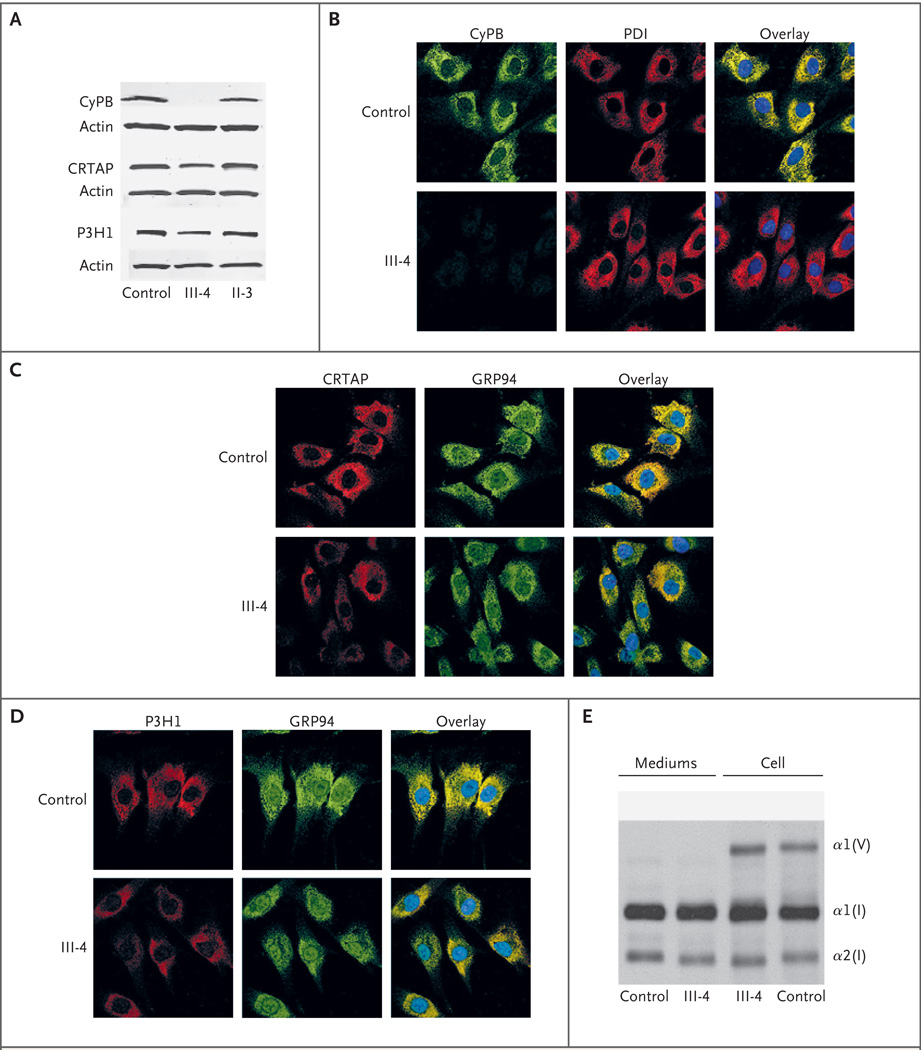
Osteogenesis imperfecta is a heritable disorder that causes bone fragility. Mutations in type I collagen result in autosomal dominant osteogenesis imperfecta, whereas mutations in either of two components of the collagen prolyl 3-hydroxylation complex (cartilage-associated protein [CRTAP] and prolyl 3-hydroxylase 1 [P3H1]) cause autosomal recessive osteogenesis imperfecta with rhizomelia (shortening of proximal segments of upper and lower limbs) and delayed collagen folding. We identified two siblings who had recessive osteogenesis imperfecta without rhizomelia. They had a homozygous start-codon mutation in the peptidyl-prolyl isomerase B gene (PPIB), which results in a lack of cyclophilin B (CyPB), the third component of the complex. The proband's collagen had normal collagen folding and normal prolyl 3-hydroxylation, suggesting that CyPB is not the exclusive peptidyl-prolyl cis-trans isomerase that catalyzes the rate-limiting step in collagen folding, as is currently thought.
2010 Massachusetts Medical Society
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
Comment in
-
Osteogenesis imperfecta, normal collagen folding, and lack of cyclophilin B.N Engl J Med. 2010 May 20;362(20):1940-1; author reply 1941-2. doi: 10.1056/NEJMc1002797. N Engl J Med. 2010. PMID: 20484404 No abstract available.
References
-
- Marini JC. Osteogenesis imperfecta. In: Behrman RE, Kliegman RM, Jensen HB, editors. Nelson textbook of pediatrics. 17th ed. Philadelphia: W.B. Saunders; 2004. pp. 2336–2338.
-
- Marini JC, Cabral WA, Barnes AM, Chang W. Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle. 2007;6:1675–1681. - PubMed
-
- Raghunath M, Bruckner P, Steinmann B. Delayed triple helix formation of mutant collagen from patients with osteogenesis imperfecta. J Mol Biol. 1994;236:940–949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- Z01 HD000408/ImNIH/Intramural NIH HHS/United States
- AR37318/AR/NIAMS NIH HHS/United States
- Z99 HD999999/ImNIH/Intramural NIH HHS/United States
- DK-54001/DK/NIDDK NIH HHS/United States
- R01 AR036794/AR/NIAMS NIH HHS/United States
- Z01 HD008830/ImNIH/Intramural NIH HHS/United States
- R01 AR037318/AR/NIAMS NIH HHS/United States
- R37 AR036794/AR/NIAMS NIH HHS/United States
- P01 HD022657/HD/NICHD NIH HHS/United States
- R37 AR037318/AR/NIAMS NIH HHS/United States
- AR36794/AR/NIAMS NIH HHS/United States
- HD22657/HD/NICHD NIH HHS/United States
- Z01 DK054001/ImNIH/Intramural NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases