

Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210
- PMID: 20089971
- PMCID: PMC3108191
- DOI: 10.1056/NEJMoa0900158
Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210
Abstract
Background: Establishing the genetic basis of phenotypes such as skeletal dysplasia in model organisms can provide insights into biologic processes and their role in human disease.
Methods: We screened mutagenized mice and observed a neonatal lethal skeletal dysplasia with an autosomal recessive pattern of inheritance. Through genetic mapping and positional cloning, we identified the causative mutation.
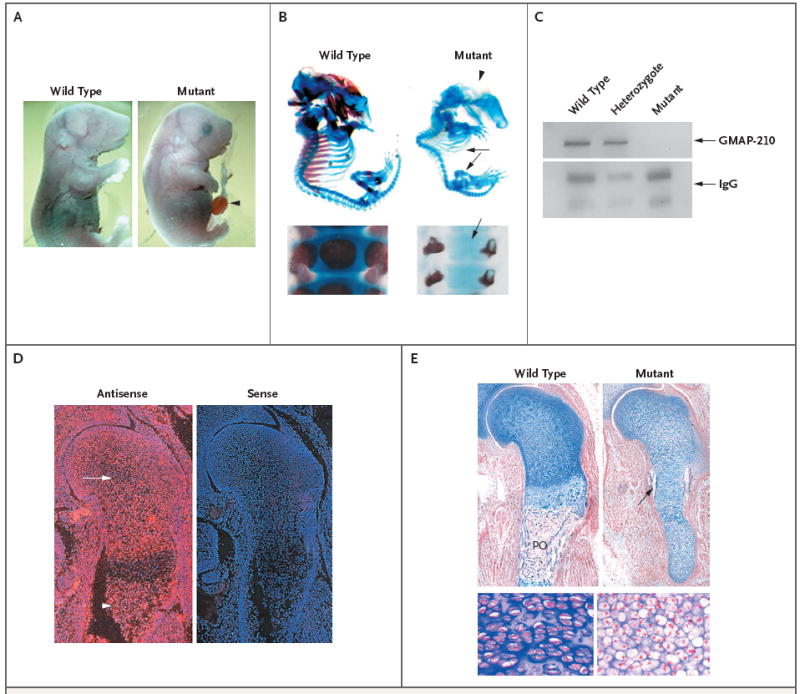
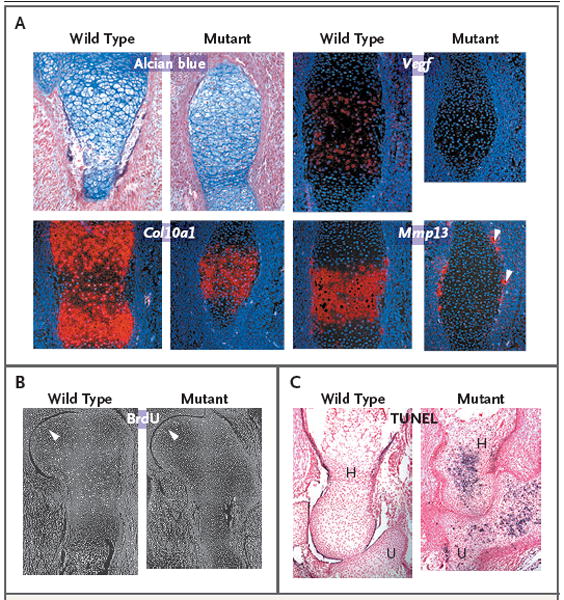
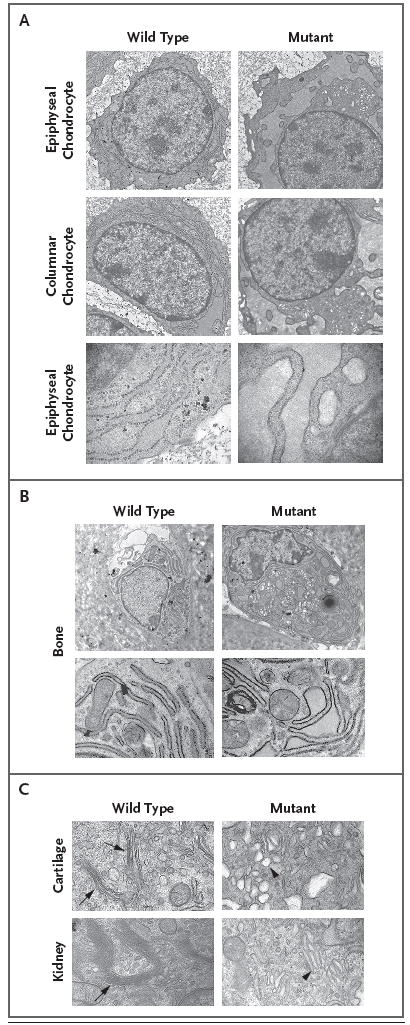
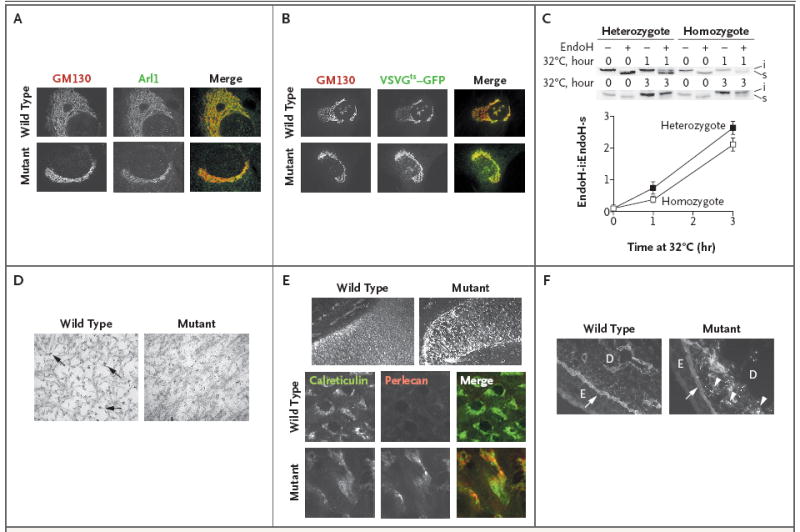
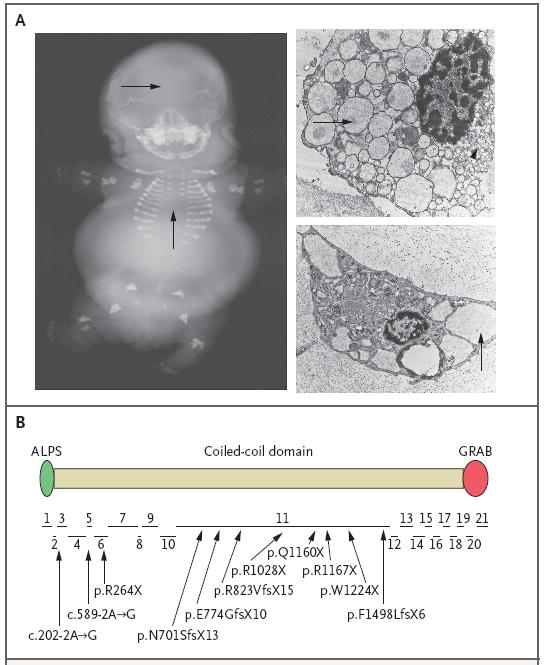
Results: Affected mice had a nonsense mutation in the thyroid hormone receptor interactor 11 gene (Trip11), which encodes the Golgi microtubule-associated protein 210 (GMAP-210); the affected mice lacked this protein. Golgi architecture was disturbed in multiple tissues, including cartilage. Skeletal development was severely impaired, with chondrocytes showing swelling and stress in the endoplasmic reticulum, abnormal cellular differentiation, and increased cell death. Golgi-mediated glycosylation events were altered in fibroblasts and chondrocytes lacking GMAP-210, and these chondrocytes had intracellular accumulation of perlecan, an extracellular matrix protein, but not of type II collagen or aggrecan, two other extracellular matrix proteins. The similarities between the skeletal and cellular phenotypes in these mice and those in patients with achondrogenesis type 1A, a neonatal lethal form of skeletal dysplasia in humans, suggested that achondrogenesis type 1A may be caused by GMAP-210 deficiency. Sequence analysis revealed loss-of-function mutations in the 10 unrelated patients with achondrogenesis type 1A whom we studied.
Conclusions: GMAP-210 is required for the efficient glycosylation and cellular transport of multiple proteins. The identification of a mutation affecting GMAP-210 in mice, and then in humans, as the cause of a lethal skeletal dysplasia underscores the value of screening for abnormal phenotypes in model organisms and identifying the causative mutations.
2010 Massachusetts Medical Society
Conflict of interest statement
No potential conflict of interest relevant to this article was reported.
Figures
Comment in
-
Achondrogenesis type 1A--from mouse to human.N Engl J Med. 2010 Jan 21;362(3):266-7. doi: 10.1056/NEJMe0911455. N Engl J Med. 2010. PMID: 20089978 No abstract available.
Similar articles
-
Achondrogenesis type 1A--from mouse to human.N Engl J Med. 2010 Jan 21;362(3):266-7. doi: 10.1056/NEJMe0911455. N Engl J Med. 2010. PMID: 20089978 No abstract available.
-
Pathogenic variants in the TRIP11 gene cause a skeletal dysplasia spectrum from odontochondrodysplasia to achondrogenesis 1A.Am J Med Genet A. 2020 Apr;182(4):681-688. doi: 10.1002/ajmg.a.61460. Epub 2020 Jan 5. Am J Med Genet A. 2020. PMID: 31903676
-
A common pathomechanism in GMAP-210- and LBR-related diseases.JCI Insight. 2018 Dec 6;3(23):e121150. doi: 10.1172/jci.insight.121150. JCI Insight. 2018. PMID: 30518689 Free PMC article.
-
Matrix composition of cartilaginous anlagen in achondrogenesis type II (Langer-Saldino).Front Biosci. 2005 Jan 1;10:446-53. doi: 10.2741/1540. Print 2005 Jan 1. Front Biosci. 2005. PMID: 15574381 Review.
-
Pseudoachondroplastic dysplasia: an Iowa review from human to mouse.Iowa Orthop J. 1999;19:53-65. Iowa Orthop J. 1999. PMID: 10847517 Free PMC article. Review.
Cited by
-
Genome-wide analysis reveals novel regulators of synaptic maintenance in Drosophila.Genetics. 2023 Apr 6;223(4):iyad025. doi: 10.1093/genetics/iyad025. Genetics. 2023. PMID: 36799927 Free PMC article.
-
A forward genetic screen with a thalamocortical axon reporter mouse yields novel neurodevelopment mutants and a distinct emx2 mutant phenotype.Neural Dev. 2011 Jan 7;6:3. doi: 10.1186/1749-8104-6-3. Neural Dev. 2011. PMID: 21214893 Free PMC article.
-
Supply chain logistics - the role of the Golgi complex in extracellular matrix production and maintenance.J Cell Sci. 2022 Jan 1;135(1):jcs258879. doi: 10.1242/jcs.258879. Epub 2022 Jan 13. J Cell Sci. 2022. PMID: 35023559 Free PMC article. Review.
-
Membrane trafficking in health and disease.Dis Model Mech. 2020 Apr 30;13(4):dmm043448. doi: 10.1242/dmm.043448. Dis Model Mech. 2020. PMID: 32433026 Free PMC article. Review.
-
The centrosome-Golgi apparatus nexus.Philos Trans R Soc Lond B Biol Sci. 2014 Sep 5;369(1650):20130462. doi: 10.1098/rstb.2013.0462. Philos Trans R Soc Lond B Biol Sci. 2014. PMID: 25047616 Free PMC article. Review.
References
-
- Barr FA, Short B. Golgins in the structure and dynamics of the Golgi apparatus. Curr Opin Cell Biol. 2003;15:405–13. - PubMed
-
- Sztul E, Lupashin V. Role of tethering factors in secretory membrane traffic. Am J Physiol Cell Physiol. 2006;290:C11–C26. - PubMed
-
- Molz G, Spycher MA. Achondrogenesis type I: light and electron-microscopic studies. Eur J Pediatr. 1980;134:69–74. - PubMed
-
- Herron BJ, Lu W, Rao C, et al. Efficient generation and mapping of recessive developmental mutations using ENU mutagenesis. Nat Genet. 2002;30:185–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases