

Within-host evolution of Burkholderia pseudomallei in four cases of acute melioidosis
- PMID: 20090837
- PMCID: PMC2799673
- DOI: 10.1371/journal.ppat.1000725
Within-host evolution of Burkholderia pseudomallei in four cases of acute melioidosis
Abstract
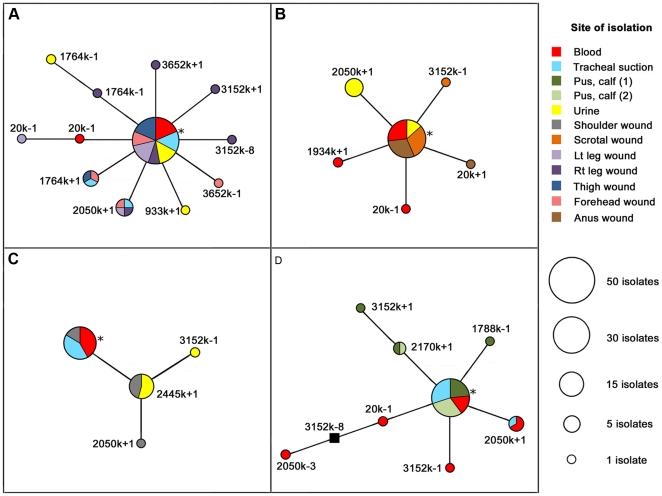
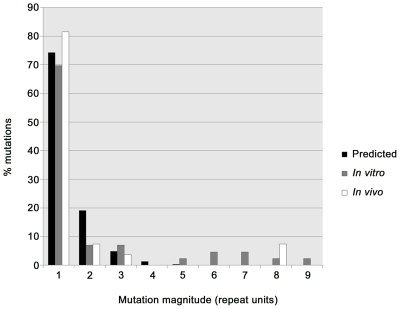
Little is currently known about bacterial pathogen evolution and adaptation within the host during acute infection. Previous studies of Burkholderia pseudomallei, the etiologic agent of melioidosis, have shown that this opportunistic pathogen mutates rapidly both in vitro and in vivo at tandemly repeated loci, making this organism a relevant model for studying short-term evolution. In the current study, B. pseudomallei isolates cultured from multiple body sites from four Thai patients with disseminated melioidosis were subjected to fine-scale genotyping using multilocus variable-number tandem repeat analysis (MLVA). In order to understand and model the in vivo variable-number tandem repeat (VNTR) mutational process, we characterized the patterns and rates of mutations in vitro through parallel serial passage experiments of B. pseudomallei. Despite the short period of infection, substantial divergence from the putative founder genotype was observed in all four melioidosis cases. This study presents a paradigm for examining bacterial evolution over the short timescale of an acute infection. Further studies are required to determine whether the mutational process leads to phenotypic alterations that impact upon bacterial fitness in vivo. Our findings have important implications for future sampling strategies, since colonies in a single clinical sample may be genetically heterogeneous, and organisms in a culture taken late in the infective process may have undergone considerable genetic change compared with the founder inoculum.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Identification of melioidosis outbreak by multilocus variable number tandem repeat analysis.Emerg Infect Dis. 2009 Feb;15(2):169-74. doi: 10.3201/eid1502.081036. Emerg Infect Dis. 2009. PMID: 19193259 Free PMC article.
-
Evolution of Burkholderia pseudomallei in recurrent melioidosis.PLoS One. 2012;7(5):e36507. doi: 10.1371/journal.pone.0036507. Epub 2012 May 15. PLoS One. 2012. PMID: 22615773 Free PMC article.
-
Validation of ten new polymorphic tandem repeat loci and application to the MLVA typing of Burkholderia pseudomallei isolates collected in Singapore from 1988 to 2004.J Microbiol Methods. 2009 Jun;77(3):297-301. doi: 10.1016/j.mimet.2009.03.005. Epub 2009 Mar 25. J Microbiol Methods. 2009. PMID: 19327380
-
Advances and remaining uncertainties in the epidemiology of Burkholderia pseudomallei and melioidosis.Trans R Soc Trop Med Hyg. 2008 Mar;102(3):225-7. doi: 10.1016/j.trstmh.2007.11.005. Epub 2007 Dec 31. Trans R Soc Trop Med Hyg. 2008. PMID: 18166205 Review.
-
The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease?FEMS Microbiol Rev. 2009 Nov;33(6):1079-99. doi: 10.1111/j.1574-6976.2009.00189.x. Epub 2009 Aug 5. FEMS Microbiol Rev. 2009. PMID: 19732156 Review.
Cited by
-
Identification of sRNA mediated responses to nutrient depletion in Burkholderia pseudomallei.Sci Rep. 2017 Dec 7;7(1):17173. doi: 10.1038/s41598-017-17356-4. Sci Rep. 2017. PMID: 29215024 Free PMC article.
-
Outcome of infection of C57BL/6 IL-10(-/-) mice with Campylobacter jejuni strains is correlated with genome content of open reading frames up- and down-regulated in vivo.Microb Pathog. 2013 Jan;54:1-19. doi: 10.1016/j.micpath.2012.08.001. Epub 2012 Aug 31. Microb Pathog. 2013. PMID: 22960579 Free PMC article.
-
Whole-genome sequencing of a quarter-century melioidosis outbreak in temperate Australia uncovers a region of low-prevalence endemicity.Microb Genom. 2016 Jul 11;2(7):e000067. doi: 10.1099/mgen.0.000067. eCollection 2016 Jul. Microb Genom. 2016. PMID: 28348862 Free PMC article.
-
Tracing melioidosis back to the source: using whole-genome sequencing to investigate an outbreak originating from a contaminated domestic water supply.J Clin Microbiol. 2015 Apr;53(4):1144-8. doi: 10.1128/JCM.03453-14. Epub 2015 Jan 28. J Clin Microbiol. 2015. PMID: 25631791 Free PMC article.
-
An objective approach for Burkholderia pseudomallei strain selection as challenge material for medical countermeasures efficacy testing.Front Cell Infect Microbiol. 2012 Sep 26;2:120. doi: 10.3389/fcimb.2012.00120. eCollection 2012. Front Cell Infect Microbiol. 2012. PMID: 23057010 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
