

Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation
- PMID: 20093275
- PMCID: PMC2871414
- DOI: 10.1074/mcp.M900516-MCP200
Post-translational modifications of integral membrane proteins resolved by top-down Fourier transform mass spectrometry with collisionally activated dissociation
Abstract
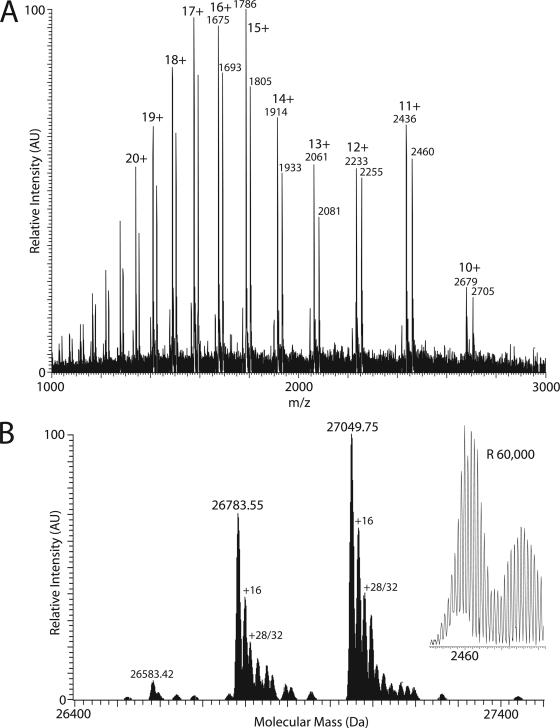
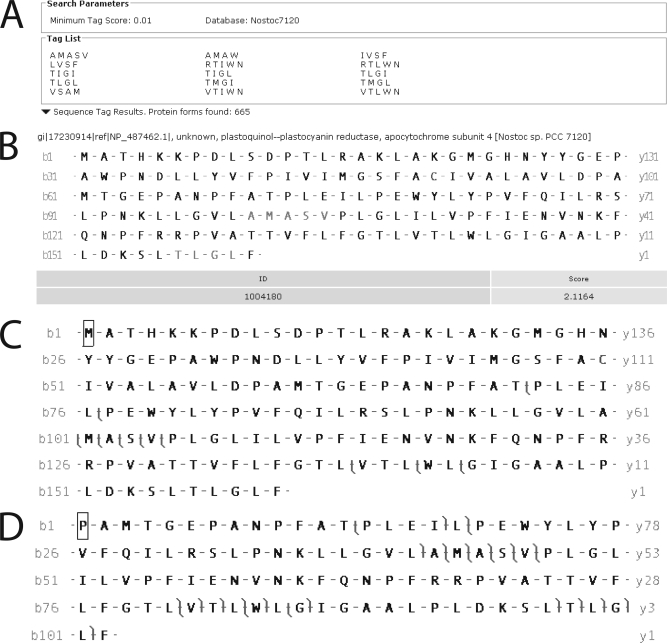
Integral membrane proteins remain a challenge to proteomics because they contain domains with physicochemical properties poorly suited to today's bottom-up protocols. These transmembrane regions may potentially contain post-translational modifications of functional significance, and thus development of protocols for improved coverage in these domains is important. One way to achieve this goal is by using top-down mass spectrometry whereby the intact protein is subjected to mass spectrometry and dissociation. Here we describe top-down high resolution Fourier transform mass spectrometry with collisionally activated dissociation to study post-translationally modified integral membrane proteins with polyhelix bundle and transmembrane porin motifs and molecular masses up to 35 kDa. On-line LC-MS analysis of the bacteriorhodopsin holoprotein yielded b- and y-ions that covered the full sequence of the protein and cleaved 79 of 247 peptide bonds (32%). The experiment proved that the mature sequence consists of residues 14-261, confirming N-terminal propeptide cleavage and conversion of N-terminal Gln-14 to pyrrolidone carboxylic acid (-17.02 Da) and C-terminal removal of Asp-262. Collisionally activated dissociation fragments localized the N(6)-(retinylidene) modification (266.20 Da) between residues 225-248 at Lys-229, the sole available amine in this stretch. Off-line nanospray of all eight subunits of the cytochrome b(6)f complex from the cyanobacterium Nostoc PCC 7120 defined various post-translational modifications, including covalently attached c-hemes (615.17 Da) on cytochromes f and b. Analysis of murine mitochondrial voltage-dependent anion channel established the amenability of the transmembrane beta-barrel to top-down MS and localized a modification site of the inhibitor Ro 68-3400 at Cys-232. Where neutral loss of the modification is a factor, only product ions that carry the modification should be used to assign its position. Although bond cleavage in some transmembrane alpha-helical domains was efficient, other regions were refractory such that their primary structure could only be inferred from the coincidence of genomic translation with precursor and product ions that spanned them.
Figures



Similar articles
-
Profiling of integral membrane proteins and their post translational modifications using high-resolution mass spectrometry.Methods. 2011 Dec;55(4):330-6. doi: 10.1016/j.ymeth.2011.09.019. Epub 2011 Sep 29. Methods. 2011. PMID: 21982782 Free PMC article. Review.
-
Sequencing covalent modifications of membrane proteins.J Exp Bot. 2006;57(7):1515-22. doi: 10.1093/jxb/erj163. Epub 2006 Mar 30. J Exp Bot. 2006. PMID: 16574746
-
Native Top-Down Mass Spectrometry Characterization of Model Integral Membrane Protein Bacteriorhodopsin.J Am Soc Mass Spectrom. 2025 May 7;36(5):961-968. doi: 10.1021/jasms.4c00439. Epub 2025 Apr 15. J Am Soc Mass Spectrom. 2025. PMID: 40234026
-
Extended Range Proteomic Analysis (ERPA): a new and sensitive LC-MS platform for high sequence coverage of complex proteins with extensive post-translational modifications-comprehensive analysis of beta-casein and epidermal growth factor receptor (EGFR).J Proteome Res. 2005 Jul-Aug;4(4):1155-70. doi: 10.1021/pr050113n. J Proteome Res. 2005. PMID: 16083266
-
Top-down mass spectrometry of integral membrane proteins.Expert Rev Proteomics. 2006 Dec;3(6):585-96. doi: 10.1586/14789450.3.6.585. Expert Rev Proteomics. 2006. PMID: 17181473 Review.
Cited by
-
Top Down proteomics: facts and perspectives.Biochem Biophys Res Commun. 2014 Mar 21;445(4):683-93. doi: 10.1016/j.bbrc.2014.02.041. Epub 2014 Feb 17. Biochem Biophys Res Commun. 2014. PMID: 24556311 Free PMC article. Review.
-
Lamina-associated domains: peripheral matters and internal affairs.Genome Biol. 2020 Apr 2;21(1):85. doi: 10.1186/s13059-020-02003-5. Genome Biol. 2020. PMID: 32241294 Free PMC article. Review.
-
Large-scale top-down proteomics of the human proteome: membrane proteins, mitochondria, and senescence.Mol Cell Proteomics. 2013 Dec;12(12):3465-73. doi: 10.1074/mcp.M113.030114. Epub 2013 Sep 10. Mol Cell Proteomics. 2013. PMID: 24023390 Free PMC article.
-
Top-down proteomics reveals concerted reductions in myofilament and Z-disc protein phosphorylation after acute myocardial infarction.Mol Cell Proteomics. 2014 Oct;13(10):2752-64. doi: 10.1074/mcp.M114.040675. Epub 2014 Jun 26. Mol Cell Proteomics. 2014. PMID: 24969035 Free PMC article.
-
Three dimensional liquid chromatography coupling ion exchange chromatography/hydrophobic interaction chromatography/reverse phase chromatography for effective protein separation in top-down proteomics.Anal Chem. 2015;87(10):5363-5371. doi: 10.1021/acs.analchem.5b00657. Epub 2015 Apr 29. Anal Chem. 2015. PMID: 25867201 Free PMC article.
References
-
- Kelleher N. L., Lin H. Y., Valaskovic G. A., Aaserud D. J., Fridriksson E. K., McLafferty F. W. (1999) Top down versus bottom up protein characterization by tandem high-resolution mass spectrometry. J. Am. Chem. Soc 121, 806–807
-
- Kelleher N. L., Zubarev R. A., Bush K., Furie B., Furie B. C., McLafferty F. W., Walsh C. T. (1999) Localization of labile posttranslational modifications by electron capture dissociation: the case of gamma-carboxyglutamic acid. Anal. Chem 71, 4250–4253 - PubMed
-
- Jebanathirajah J. A., Pittman J. L., Thomson B. A., Budnik B. A., Kaur P., Rape M., Kirschner M., Costello C. E., O'Connor P. B. (2005) Characterization of a new qQq-FTICR mass spectrometer for post-translational modification analysis and top-down tandem mass spectrometry of whole proteins. J. Am. Soc. Mass Spectrom 16, 1985–1999 - PubMed
-
- Wu C. C., MacCoss M. J., Howell K. E., Yates J. R., 3rd (2003) A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol 21, 532–538 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
