

The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients
- PMID: 20093774
- PMCID: PMC2810088
- DOI: 10.1172/JCI40706
The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients
Abstract
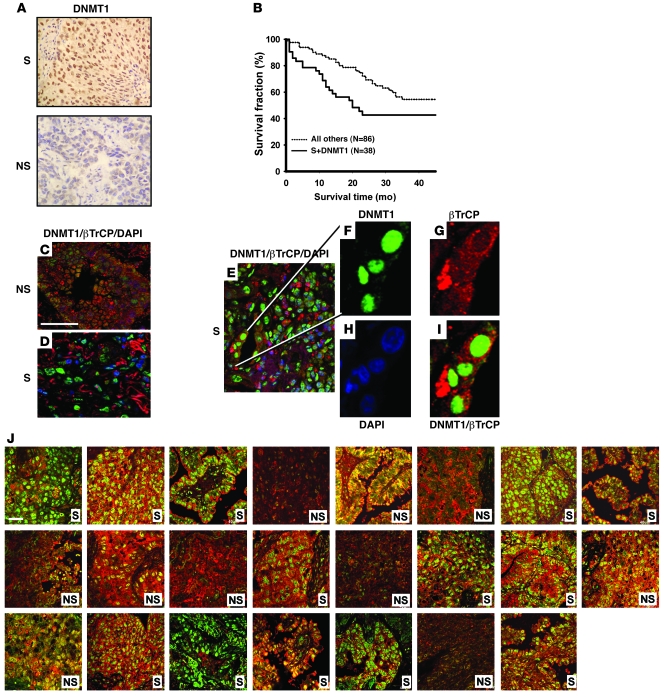
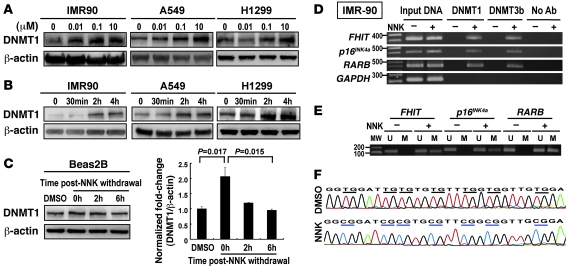
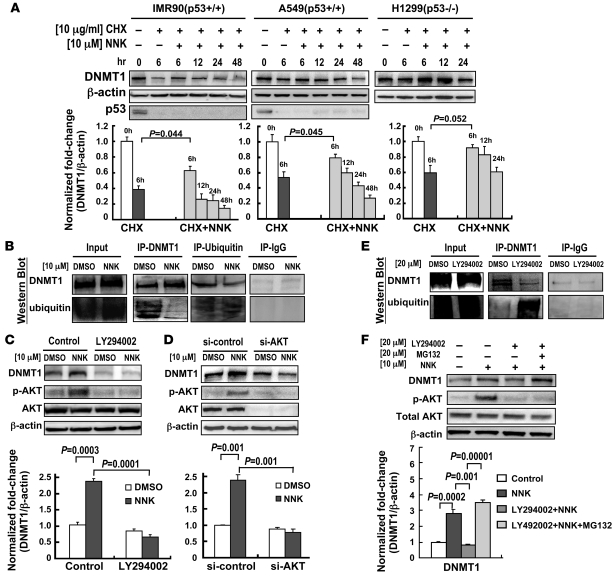
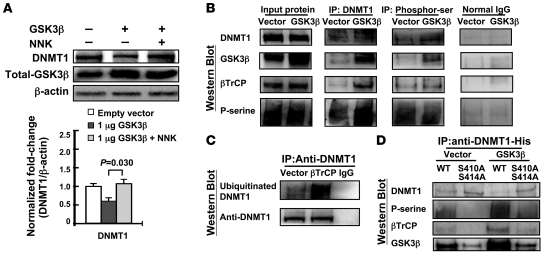
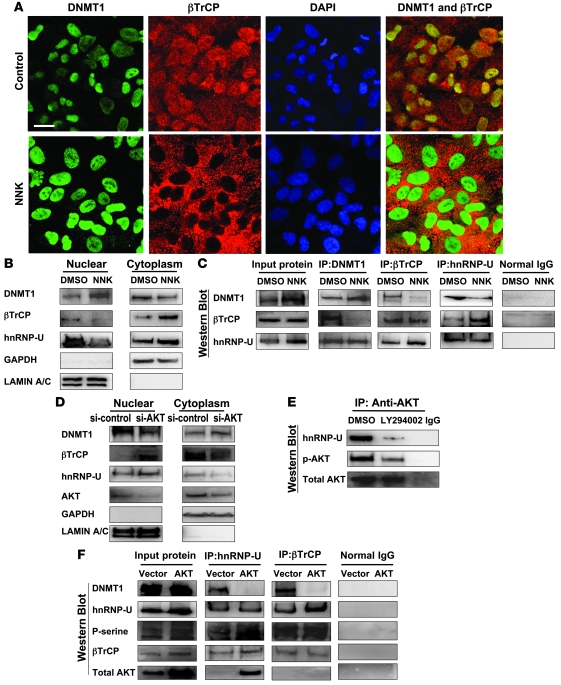
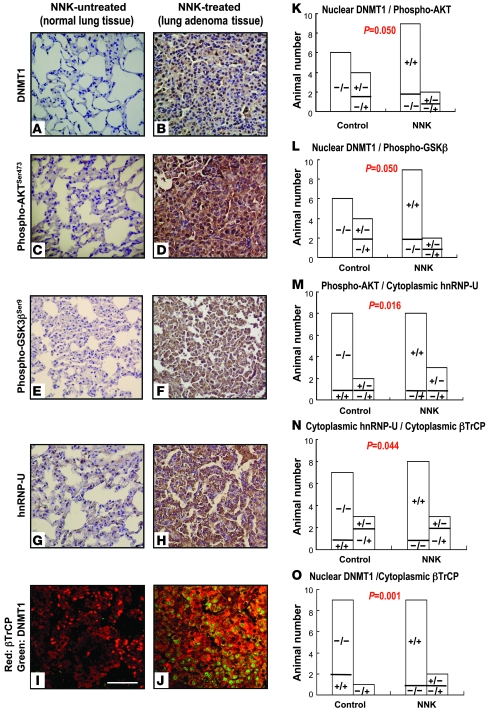
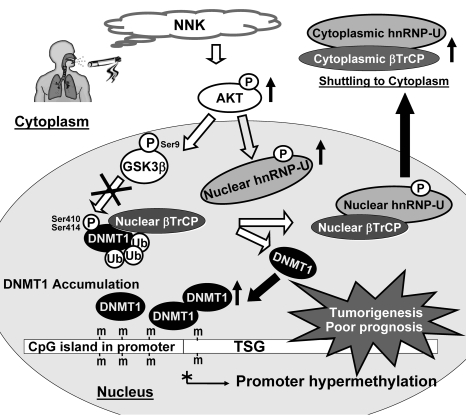
DNA methyltransferase 1 (DNMT1) catalyzes DNA methylation and is overexpressed in many human diseases, including cancer. The tobacco-specific carcinogen NNK also induces DNA methylation. However, the role of DNMT1-mediated methylation in tobacco carcinogenesis remains unclear. Here we used human and mouse lung cancer samples and cell lines to determine a mechanism whereby NNK induced DNMT1 expression and activity. We determined that in a human lung cell line, glycogen synthase kinase 3beta (GSK3beta) phosphorylated DNMT1 to recruit beta-transducin repeat-containing protein (betaTrCP), resulting in DNMT1 degradation, and that NNK activated AKT, inhibiting GSK3beta function and thereby attenuating DNMT1 degradation. NNK also induced betaTrCP translocation to the cytoplasm via the heterogeneous nuclear ribonucleoprotein U (hnRNP-U) shuttling protein, resulting in DNMT1 nuclear accumulation and hypermethylation of the promoters of tumor suppressor genes. Fluorescence immunohistochemistry (IHC) of lung adenomas from NNK-treated mice and tumors from lung cancer patients that were smokers were characterized by disruption of the DNMT1/betaTrCP interaction and DNMT1 nuclear accumulation. Importantly, DNMT1 overexpression in lung cancer patients who smoked continuously correlated with poor prognosis. We believe that the NNK-induced DNMT1 accumulation and subsequent hypermethylation of the promoter of tumor suppressor genes may lead to tumorigenesis and poor prognosis and provide an important link between tobacco smoking and lung cancer. Furthermore, this mechanism may also be involved in other smoking-related human diseases.
Figures
References
-
- Costa E, Dong E, Grayson DR, Guidotti A, Ruzicka W, Veldic M. Reviewing the role of DNA (cytosine-5) methyltransferase overexpression in the cortical GABAergic dysfunction associated with psychosis vulnerability. Epigenetics. 2007;2(1):29–36. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
