

Genetics and molecular pathology of Stargardt-like macular degeneration
- PMID: 20096366
- PMCID: PMC3059896
- DOI: 10.1016/j.preteyeres.2010.01.001
Genetics and molecular pathology of Stargardt-like macular degeneration
Abstract
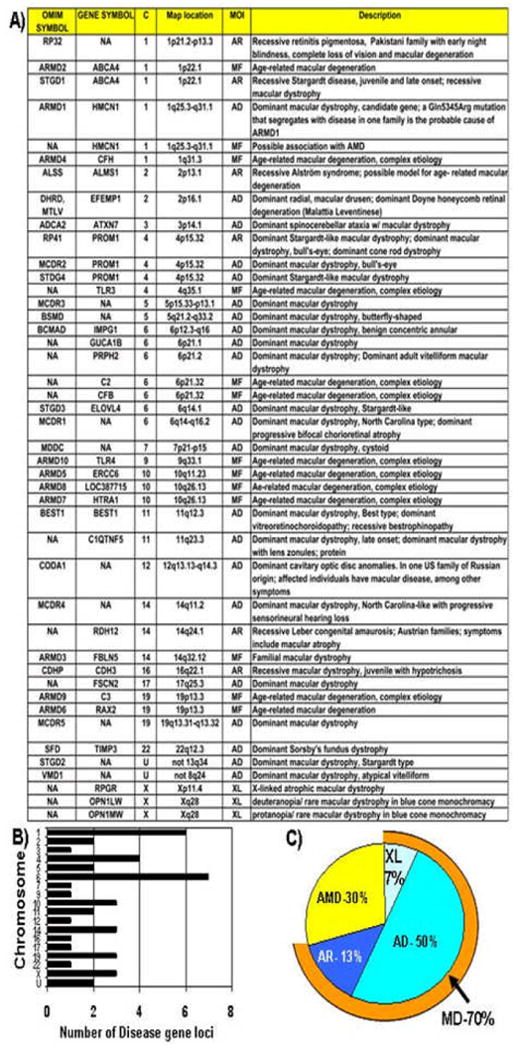
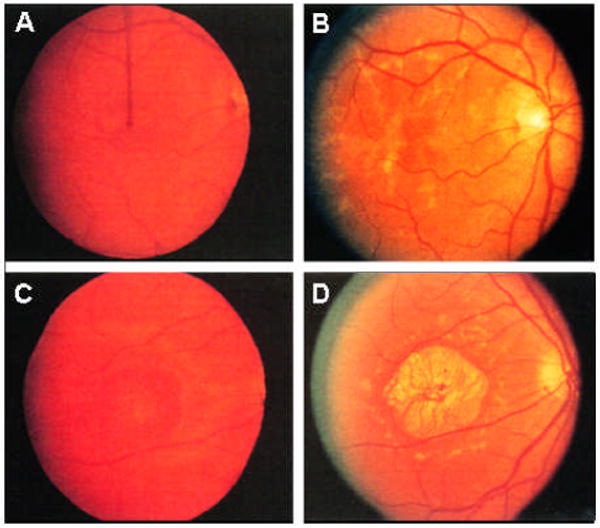
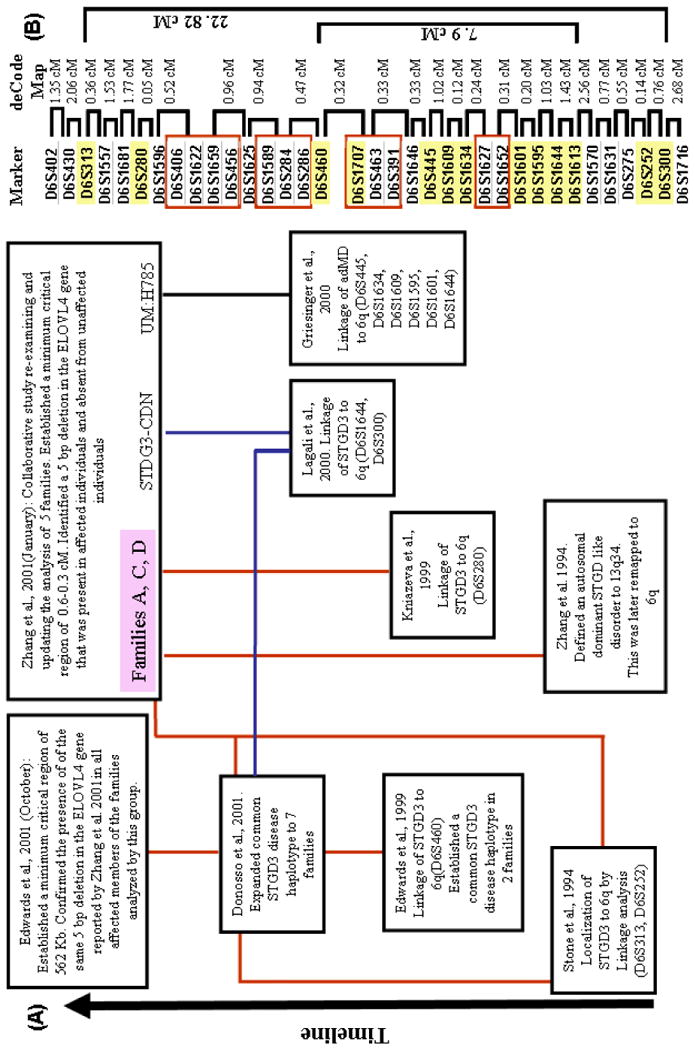
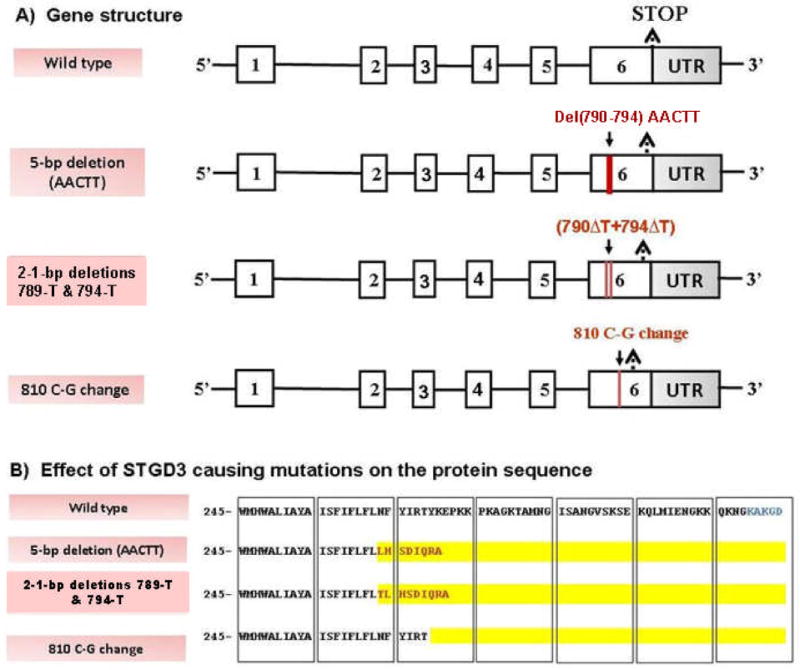
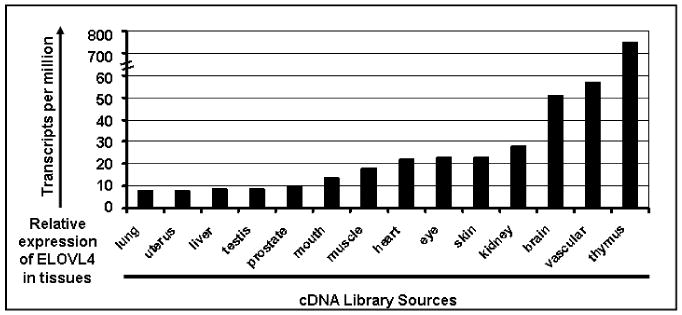
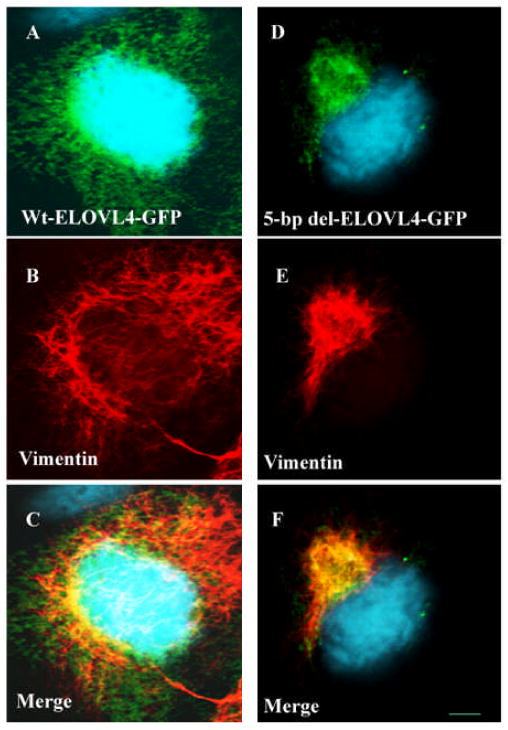
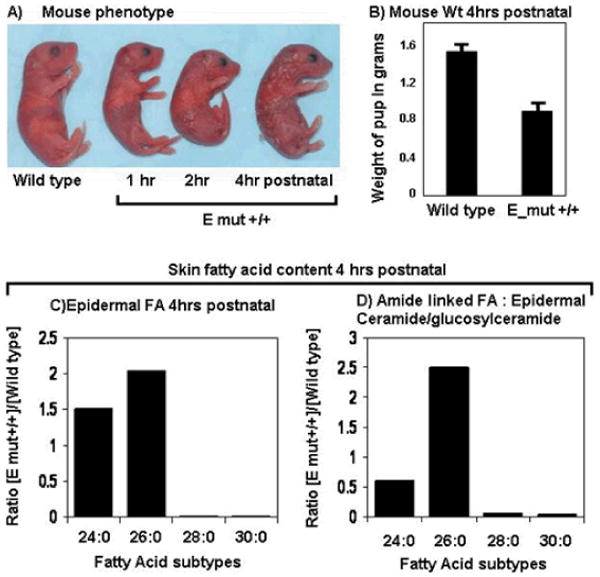
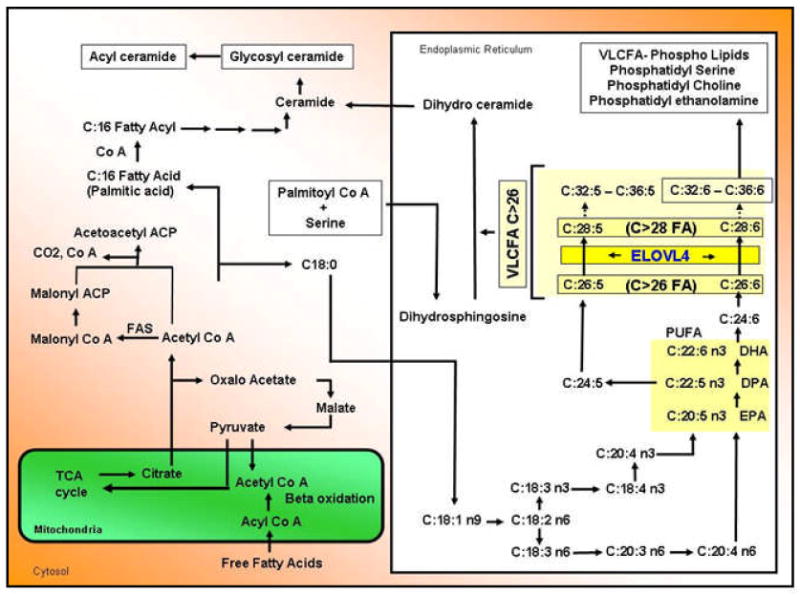
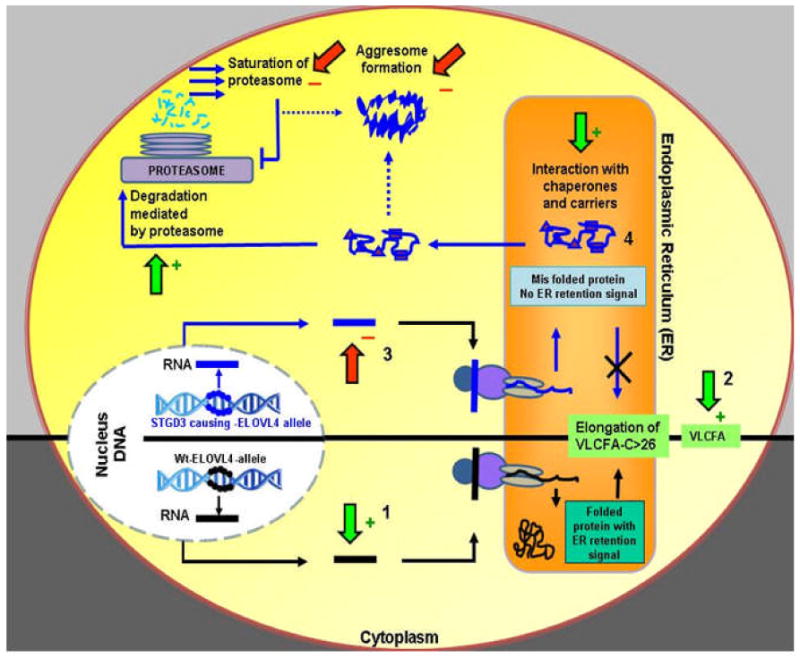
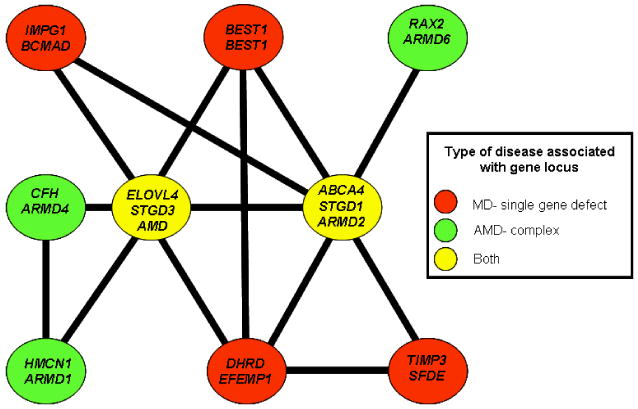
Stargardt-like macular degeneration (STGD3) is an early onset, autosomal dominant macular degeneration. STGD3 is characterized by a progressive pathology, the loss of central vision, atrophy of the retinal pigment epithelium, and accumulation of lipofuscin, clinical features that are also characteristic of age-related macular degeneration. The onset of clinical symptoms in STGD3, however, is typically observed within the second or third decade of life (i.e., starting in the teenage years). The clinical profile at any given age among STGD3 patients can be variable suggesting that, although STGD3 is a single gene defect, other genetic or environmental factors may play a role in moderating the final disease phenotype. Genetic studies localized the STGD3 disease locus to a small region on the short arm of human chromosome 6, and application of a positional candidate gene approach identified protein truncating mutations in the elongation of very long chain fatty acids-4 gene (ELOVL4) in patients with this disease. The ELOVL4 gene encodes a protein homologous to the ELO group of proteins that participate in fatty acid elongation in yeast. Pathogenic mutations found in the ELOVL4 gene result in altered trafficking of the protein and behave with a dominant negative effect. Mice carrying an Elovl4 mutation developed photoreceptor degeneration and depletion of very long chain fatty acids (VLCFA). ELOVL4 protein participates in the synthesis of fatty acids with chain length longer than 26 carbons. Studies on ELOVL4 indicate that VLCFA may be necessary for normal function of the retina, and the defective protein trafficking and/or altered VLCFA elongation underlies the pathology associated with STGD3. Determining the role of VLCFA in the retina and discerning the implications of abnormal trafficking of mutant ELOVL4 and depleted VLCFA content in the pathology of STGD3 will provide valuable insight in understanding the retinal structure, function, and pathology underlying STGD3 and may lead to a better understanding of the process of macular disease in general.
(c) 2010. Published by Elsevier Ltd.
Figures
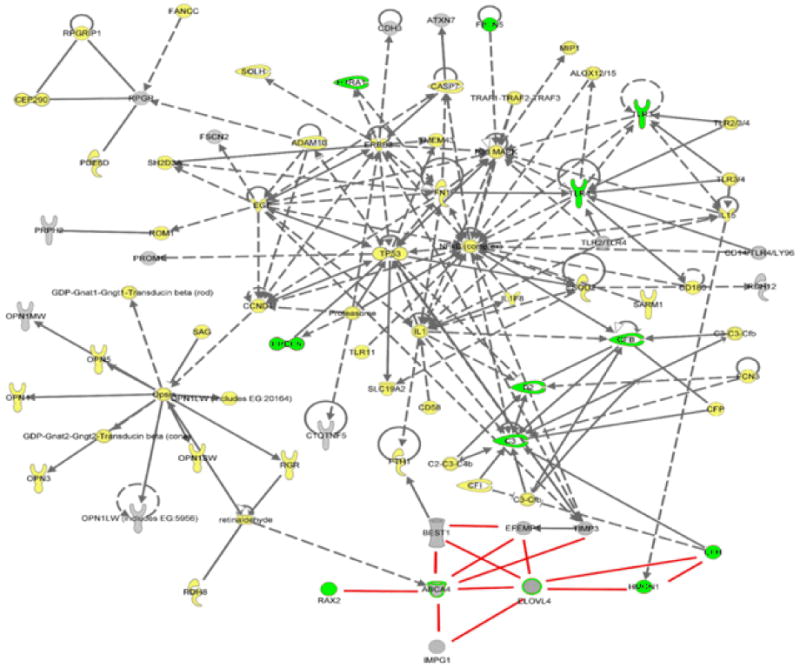
| Cytokine/growth factor |  | Ion channel |  |
| Chemical/toxicant |  | Peptidase |  |
| Enzyme |  | Transcription regulator |  |
| G-protein coupled receptor |  | Transmembrane receptor |  |
| Group/complex/ |  | other transporter |  |
| Growth factor |  | Undefined |  |
References
-
- Agbaga MP, Mandal MA, Brush RS, Zheng L, Henry K, Elliott MH, Vasireddy V, Petrukhin K, Ayyagari R, Anderson RE. Quantitative analysis of ELOVL4 protein and fatty acid products in knock-out and knock-in mouse tissues. The Association for Research in Vision and Ophthalmology; Fort Lauderdale, Florida, USA: 2009.
-
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. - PubMed
-
- Ambasudhan R, Wang X, Jablonski MM, Thompson DA, Lagali PS, Wong PW, Sieving PA, Ayyagari R. Atrophic macular degeneration mutations in ELOVL4 result in the intracellular misrouting of the protein. Genomics. 2004;83:615–625. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
