

Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering
- PMID: 20097063
- PMCID: PMC2829847
- DOI: 10.1016/j.sbi.2009.12.015
Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering
Abstract
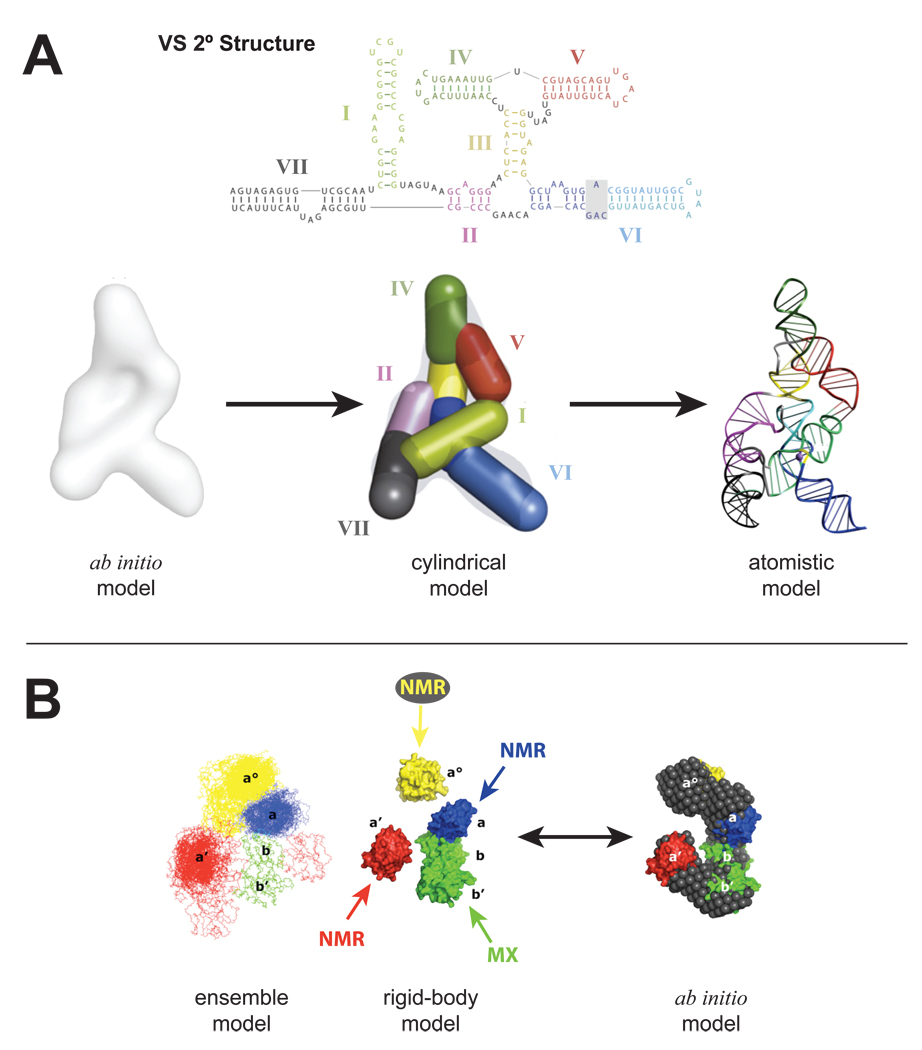
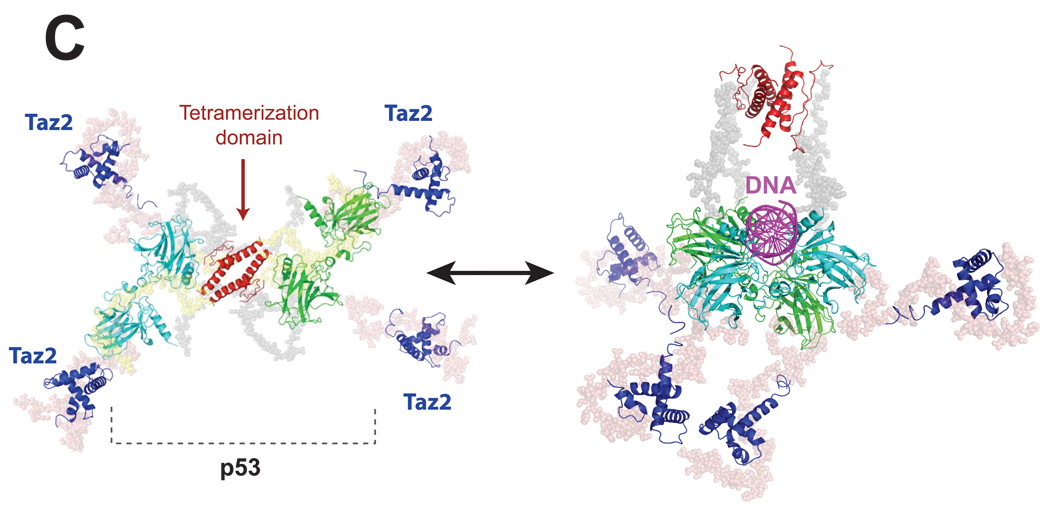
Small-angle X-ray scattering (SAXS) is changing how we perceive biological structures, because it reveals dynamic macromolecular conformations and assemblies in solution. SAXS information captures thermodynamic ensembles, enhances static structures detailed by high-resolution methods, uncovers commonalities among diverse macromolecules, and helps define biological mechanisms. SAXS-based experiments on RNA riboswitches and ribozymes and on DNA-protein complexes including DNA-PK and p53 discover flexibilities that better define structure-function relationships. Furthermore, SAXS results suggest conformational variation is a general functional feature of macromolecules. Thus, accurate structural analyses will require a comprehensive approach that assesses both flexibility, as seen by SAXS, and detail, as determined by X-ray crystallography and NMR. Here, we review recent SAXS computational tools, technologies, and applications to nucleic acids and related structures.
Published by Elsevier Ltd.
Figures



References
-
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. - PubMed
-
- Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. - PubMed
-
-
Hura GL, Menon AL, Hammel M, Rambo RP, Poole FL, 2nd, Tsutakawa SE, Jenney FE, Jr, Classen S, Frankel KA, Hopkins RC, et al. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS) Nat Methods. 2009;6:606–612.. This article presents an automated system and data analysis pipeline for SAXS data collection. The results on 50 samples indicate the relatively small amounts of material needed, the robust features of SAXS for efficient throughput, and the high percentage of samples for which structural data can be obtained.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
