

Two-dimensional NMR and all-atom molecular dynamics of cytochrome P450 CYP119 reveal hidden conformational substates
- PMID: 20097757
- PMCID: PMC2843209
- DOI: 10.1074/jbc.M109.087593
Two-dimensional NMR and all-atom molecular dynamics of cytochrome P450 CYP119 reveal hidden conformational substates
Abstract
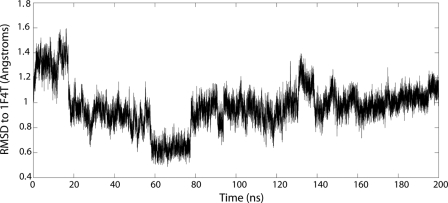
Cytochrome P450 enzymes are versatile catalysts involved in a wide variety of biological processes from hormonal regulation and antibiotic synthesis to drug metabolism. A hallmark of their versatility is their promiscuous nature, allowing them to recognize a wide variety of chemically diverse substrates. However, the molecular details of this promiscuity have remained elusive. Here, we have utilized two-dimensional heteronuclear single quantum coherence NMR spectroscopy to examine a series of mutants site-specific labeled with the unnatural amino acid, [(13)C]p-methoxyphenylalanine, in conjunction with all-atom molecular dynamics simulations to examine substrate and inhibitor binding to CYP119, a P450 from Sulfolobus acidocaldarius. The results suggest that tight binding hydrophobic ligands tend to lock the enzyme into a single conformational substate, whereas weak binding low affinity ligands bind loosely in the active site, resulting in a distribution of localized conformers. Furthermore, the molecular dynamics simulations suggest that the ligand-free enzyme samples ligand-bound conformations of the enzyme and, therefore, that ligand binding may proceed largely through a process of conformational selection rather than induced fit.
Figures






Similar articles
-
Analysis of cytochrome P450 CYP119 ligand-dependent conformational dynamics by two-dimensional NMR and X-ray crystallography.J Biol Chem. 2015 Apr 17;290(16):10000-17. doi: 10.1074/jbc.M114.627935. Epub 2015 Feb 10. J Biol Chem. 2015. PMID: 25670859 Free PMC article.
-
Ligand-induced conformational heterogeneity of cytochrome P450 CYP119 identified by 2D NMR spectroscopy with the unnatural amino acid (13)C-p-methoxyphenylalanine.J Am Chem Soc. 2008 Dec 3;130(48):16168-9. doi: 10.1021/ja8071463. J Am Chem Soc. 2008. PMID: 18998650 Free PMC article.
-
Regioselective oxidation of lauric acid by CYP119, an orphan cytochrome P450 from Sulfolobus acidocaldarius.J Microbiol Biotechnol. 2010 Mar;20(3):574-8. J Microbiol Biotechnol. 2010. PMID: 20372030
-
Structural features of cytochromes P450 and ligands that affect drug metabolism as revealed by X-ray crystallography and NMR.Future Med Chem. 2010 Sep;2(9):1451-68. doi: 10.4155/fmc.10.229. Future Med Chem. 2010. PMID: 21103389 Free PMC article. Review.
-
Cytochrome P450 structure-function: insights from molecular dynamics simulations.Drug Metab Rev. 2016 Aug;48(3):434-52. doi: 10.1080/03602532.2016.1178771. Epub 2016 May 10. Drug Metab Rev. 2016. PMID: 27167388 Review.
Cited by
-
Stochastic ensembles, conformationally adaptive teamwork, and enzymatic detoxification.Biochemistry. 2011 May 17;50(19):3866-72. doi: 10.1021/bi200275r. Epub 2011 Apr 20. Biochemistry. 2011. PMID: 21473615 Free PMC article.
-
An efficient protocol for incorporation of an unnatural amino acid in perdeuterated recombinant proteins using glucose-based media.J Biomol NMR. 2012 Mar;52(3):191-5. doi: 10.1007/s10858-012-9606-9. Epub 2012 Feb 17. J Biomol NMR. 2012. PMID: 22350951 Free PMC article.
-
Discovery of processive catalysis by an exo-hydrolase with a pocket-shaped active site.Nat Commun. 2019 May 20;10(1):2222. doi: 10.1038/s41467-019-09691-z. Nat Commun. 2019. PMID: 31110237 Free PMC article.
-
Dynamic Effects of CYP2D6 Genetic Variants in a Set of Poor Metaboliser Patients with Infiltrating Ductal Cancer Under Treatment with Tamoxifen.Sci Rep. 2019 Feb 21;9(1):2521. doi: 10.1038/s41598-018-38340-6. Sci Rep. 2019. PMID: 30792473 Free PMC article.
-
Enhancing the efficiency and regioselectivity of P450 oxidation catalysts by unnatural amino acid mutagenesis.Chembiochem. 2014 May 5;15(7):1001-10. doi: 10.1002/cbic.201400060. Epub 2014 Apr 1. Chembiochem. 2014. PMID: 24692265 Free PMC article.
References
-
- Henzler-Wildman K. A., Lei M., Thai V., Kerns S. J., Karplus M., Kern D. (2007) Nature 450, 913–916 - PubMed
-
- Eisenmesser E. Z., Millet O., Labeikovsky W., Korzhnev D. M., Wolf-Watz M., Bosco D. A., Skalicky J. J., Kay L. E., Kern D. (2005) Nature 438, 117–121 - PubMed
-
- Tokuriki N., Tawfik D. S. (2009) Science 324, 203–207 - PubMed
-
- Benkovic S. J., Hammes-Schiffer S. (2003) Science 301, 1196–1202 - PubMed
-
- Fersht A. (1999) Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding, 2nd. Ed., pp. 54–84, W.H. Freeman and Co., New York
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
