

SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models
- PMID: 20098500
- PMCID: PMC2800046
- DOI: 10.1371/journal.pcbi.1000644
SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models
Abstract
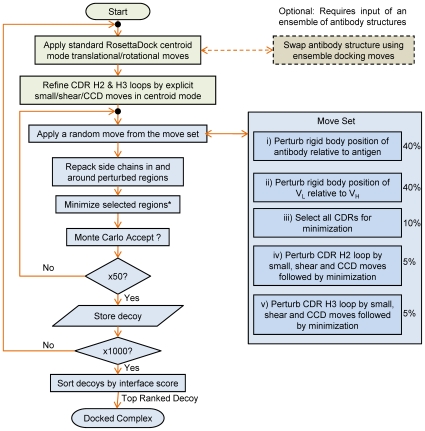
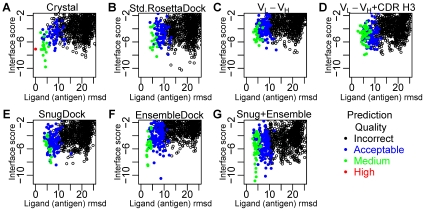
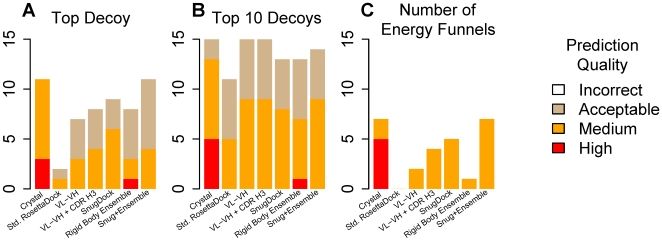
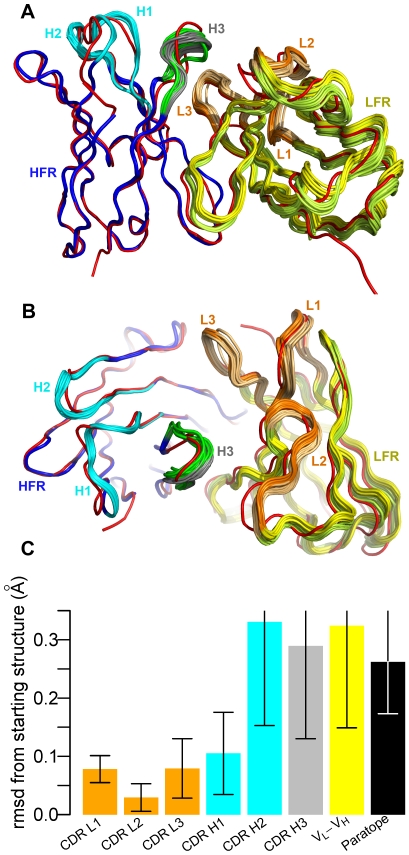
High resolution structures of antibody-antigen complexes are useful for analyzing the binding interface and to make rational choices for antibody engineering. When a crystallographic structure of a complex is unavailable, the structure must be predicted using computational tools. In this work, we illustrate a novel approach, named SnugDock, to predict high-resolution antibody-antigen complex structures by simultaneously structurally optimizing the antibody-antigen rigid-body positions, the relative orientation of the antibody light and heavy chains, and the conformations of the six complementarity determining region loops. This approach is especially useful when the crystal structure of the antibody is not available, requiring allowances for inaccuracies in an antibody homology model which would otherwise frustrate rigid-backbone docking predictions. Local docking using SnugDock with the lowest-energy RosettaAntibody homology model produced more accurate predictions than standard rigid-body docking. SnugDock can be combined with ensemble docking to mimic conformer selection and induced fit resulting in increased sampling of diverse antibody conformations. The combined algorithm produced four medium (Critical Assessment of PRediction of Interactions-CAPRI rating) and seven acceptable lowest-interface-energy predictions in a test set of fifteen complexes. Structural analysis shows that diverse paratope conformations are sampled, but docked paratope backbones are not necessarily closer to the crystal structure conformations than the starting homology models. The accuracy of SnugDock predictions suggests a new genre of general docking algorithms with flexible binding interfaces targeted towards making homology models useful for further high-resolution predictions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Toward high-resolution homology modeling of antibody Fv regions and application to antibody-antigen docking.Proteins. 2009 Feb 1;74(2):497-514. doi: 10.1002/prot.22309. Proteins. 2009. PMID: 19062174 Free PMC article.
-
Paratope states in solution improve structure prediction and docking.Structure. 2022 Mar 3;30(3):430-440.e3. doi: 10.1016/j.str.2021.11.001. Epub 2021 Nov 26. Structure. 2022. PMID: 34838187
-
A computational approach for studying antibody-antigen interactions without prior structural information: the anti-testosterone binding antibody as a case study.Proteins. 2017 Feb;85(2):322-331. doi: 10.1002/prot.25226. Epub 2016 Dec 26. Proteins. 2017. PMID: 27936519
-
Antigen-Antibody Complexes.Subcell Biochem. 2020;94:465-497. doi: 10.1007/978-3-030-41769-7_19. Subcell Biochem. 2020. PMID: 32189312 Review.
-
High-resolution protein-protein docking.Curr Opin Struct Biol. 2006 Apr;16(2):183-93. doi: 10.1016/j.sbi.2006.03.003. Epub 2006 Mar 20. Curr Opin Struct Biol. 2006. PMID: 16546374 Review.
Cited by
-
Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE).PLoS One. 2013 May 22;8(5):e63906. doi: 10.1371/journal.pone.0063906. Print 2013. PLoS One. 2013. PMID: 23717507 Free PMC article.
-
Optimal target saturation of ligand-blocking anti-GITR antibody IBI37G5 dictates FcγR-independent GITR agonism and antitumor activity.Cell Rep Med. 2022 Jun 21;3(6):100660. doi: 10.1016/j.xcrm.2022.100660. Cell Rep Med. 2022. PMID: 35732156 Free PMC article.
-
Antibody engineering and therapeutics, The Annual Meeting of the Antibody Society: December 8-12, 2013, Huntington Beach, CA.MAbs. 2014 May-Jun;6(3):577-618. doi: 10.4161/mabs.28421. Epub 2014 Mar 3. MAbs. 2014. PMID: 24589717 Free PMC article.
-
Accurate Structure Prediction of CDR H3 Loops Enabled by a Novel Structure-Based C-Terminal Constraint.J Immunol. 2017 Jan 1;198(1):505-515. doi: 10.4049/jimmunol.1601137. Epub 2016 Nov 21. J Immunol. 2017. PMID: 27872211 Free PMC article.
-
Low-resolution structural modeling of protein interactome.Curr Opin Struct Biol. 2013 Apr;23(2):198-205. doi: 10.1016/j.sbi.2012.12.003. Epub 2013 Jan 5. Curr Opin Struct Biol. 2013. PMID: 23294579 Free PMC article. Review.
References
-
- Gray JJ. High-resolution protein-protein docking. Curr Opin Struct Biol. 2006;16:183–193. - PubMed
-
- Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol. 1987;196:901–917. - PubMed
-
- Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273:927–948. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
