

The SAM domains of Anks family proteins are critically involved in modulating the degradation of EphA receptors
- PMID: 20100865
- PMCID: PMC2838079
- DOI: 10.1128/MCB.01605-09
The SAM domains of Anks family proteins are critically involved in modulating the degradation of EphA receptors
Abstract
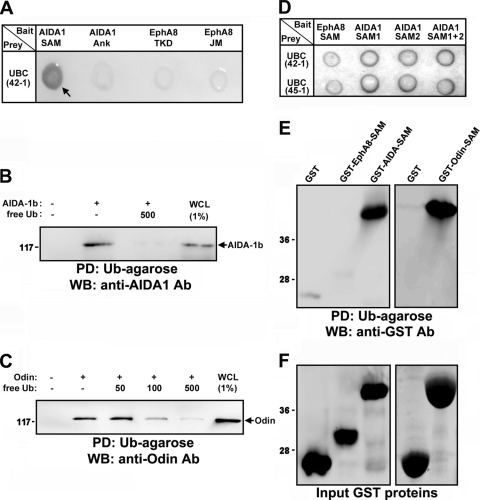
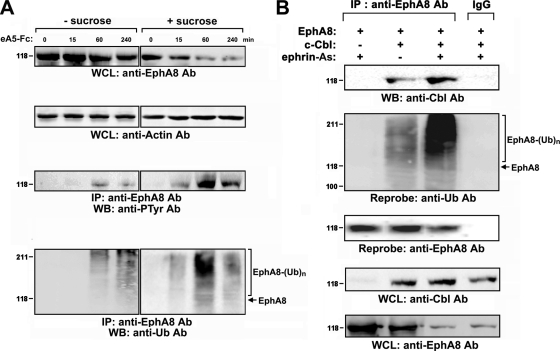
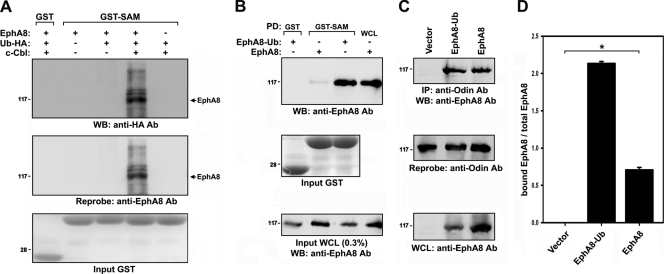
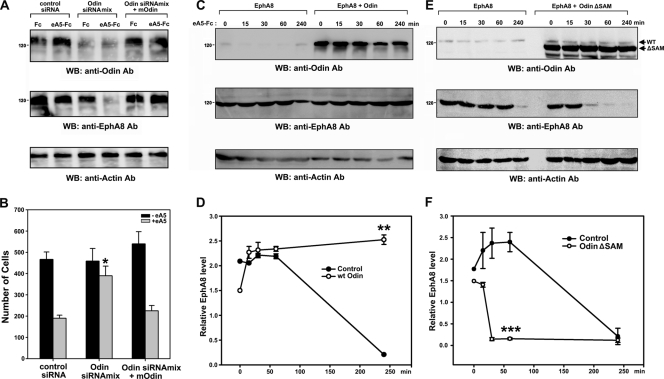
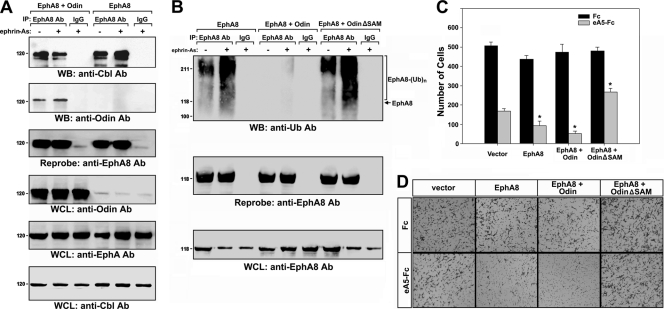
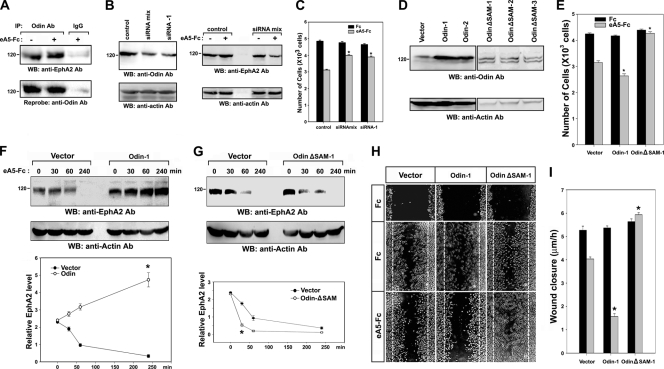
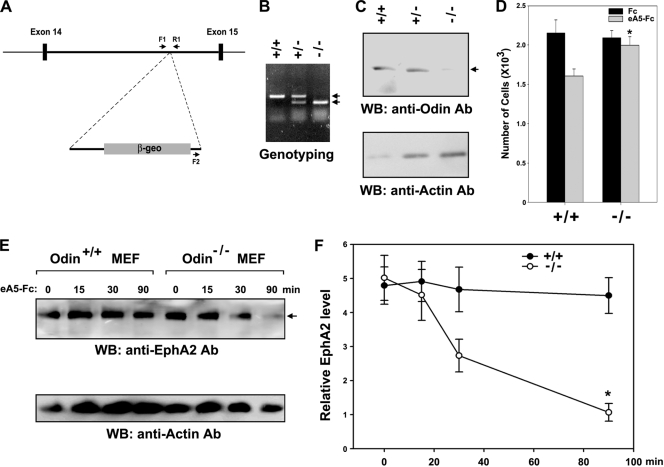
We recently reported that the phosphotyrosine-binding (PTB) domain of Anks family proteins binds to EphA8, thereby positively regulating EphA8-mediated signaling pathways. In the current study, we identified a potential role for the SAM domains of Anks family proteins in EphA signaling. We found that SAM domains of Anks family proteins directly bind to ubiquitin, suggesting that Anks proteins regulate the degradation of ubiquitinated EphA receptors. Consistent with the role of Cbl ubiquitin ligases in the degradation of Eph receptors, our results revealed that the ubiquitin ligase c-Cbl induced the ubiquitination and degradation of EphA8 upon ligand binding. Ubiquitinated EphA8 also bound to the SAM domains of Odin, a member of the Anks family proteins. More importantly, the overexpression of wild-type Odin protected EphA8 and EphA2 from undergoing degradation following ligand stimulation and promoted EphA-mediated inhibition of cell migration. In contrast, a SAM domain deletion mutant of Odin strongly impaired the function of endogenous Odin, suggesting that the mutant functions in a dominant-negative manner. An analysis of Odin-deficient primary embryonic fibroblasts indicated that Odin levels play a critical role in regulating the stability of EphA2 in response to ligand stimulation. Taken together, our studies suggest that the SAM domains of Anks family proteins play a pivotal role in enhancing the stability of EphA receptors by modulating the ubiquitination process.
Figures
References
-
- Batlle, E., J. Bacani, H. Begthel, S. Jonkheer, A. Gregorieff, M. van de Born, N. Malats, E. Sancho, E. Boon, T. Pawson, S. Gallinger, S. Pals, and H. Clevers. 2005. EphB receptor activity suppresses colorectal cancer progression. Nature 435:1126-1130. - PubMed
-
- Brantley-Sieders, D. M., G. Zhuang, D. Hicks, W. B. Fang, Y. Hwang, J. M. Cates, K. Coffman, D. Jackson, E. Bruckheimer, R. S. Muraoka-Cook, and J. Chen. 2008. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Invest. 118:64-78. - PMC - PubMed
-
- Cheng, N., D. M. Brantley, H. Liu, Q. Lin, M. Enriquez, N. Gale, G. Yancopoulos, D. P. Cerretti, T. O. Daniel, and J. Chen. 2002. Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis. Mol. Cancer Res. 1:2-11. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous