

Protein folding in membranes
- PMID: 20101433
- PMCID: PMC11115603
- DOI: 10.1007/s00018-010-0259-0
Protein folding in membranes
Abstract
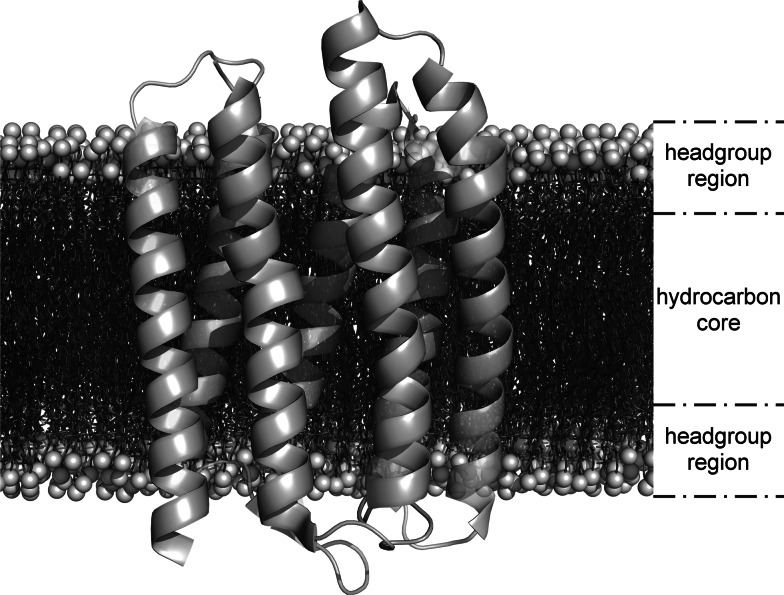
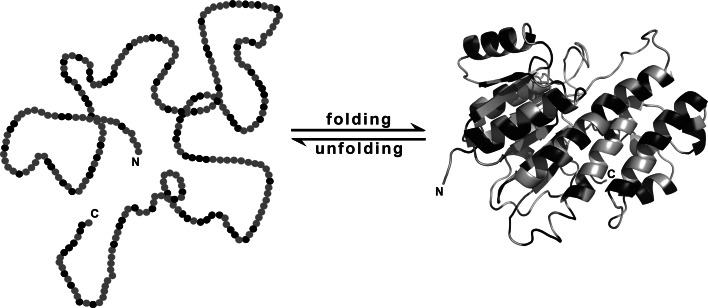
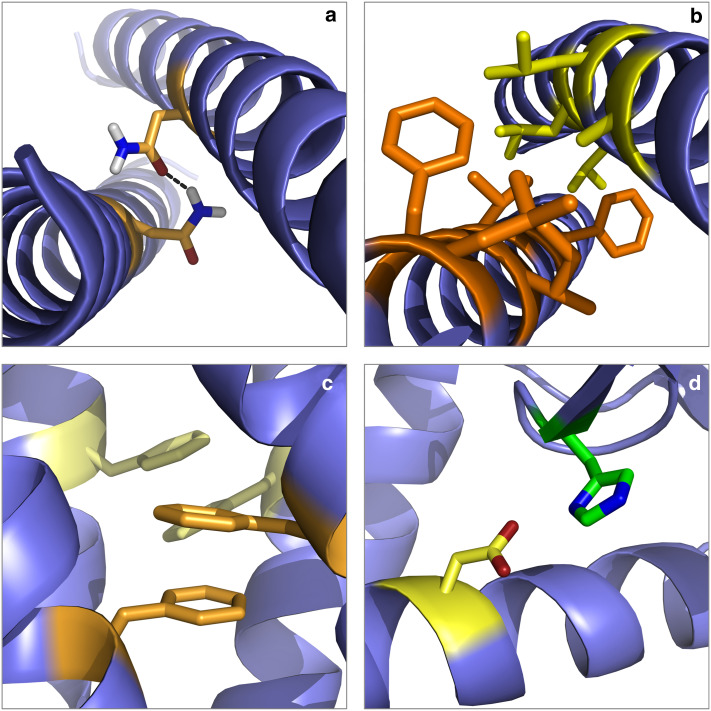
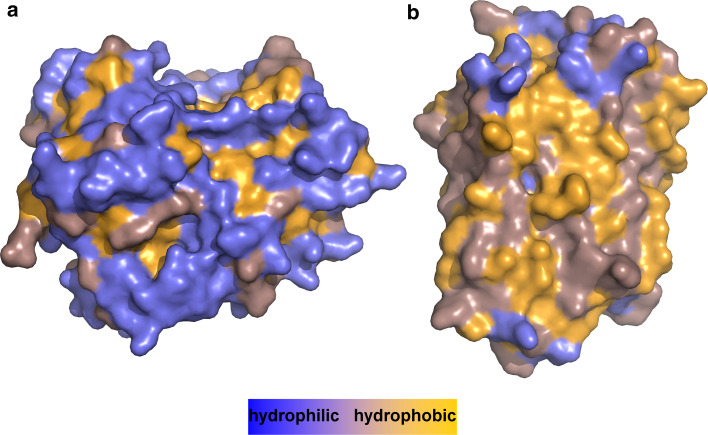
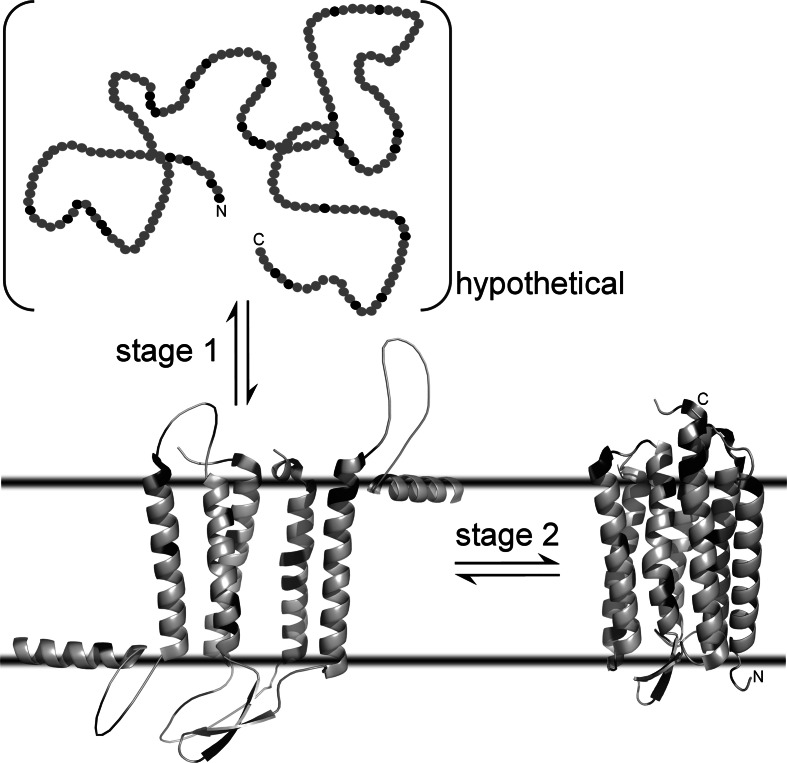
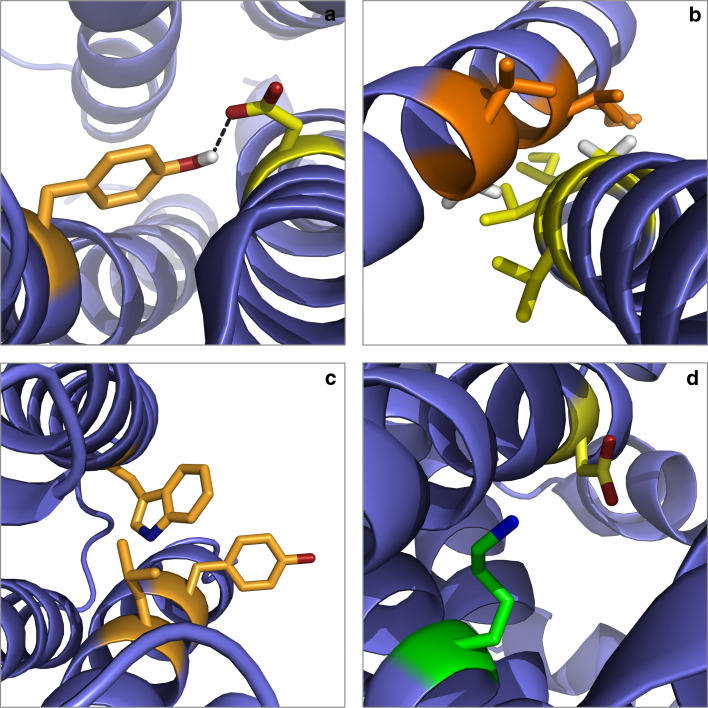
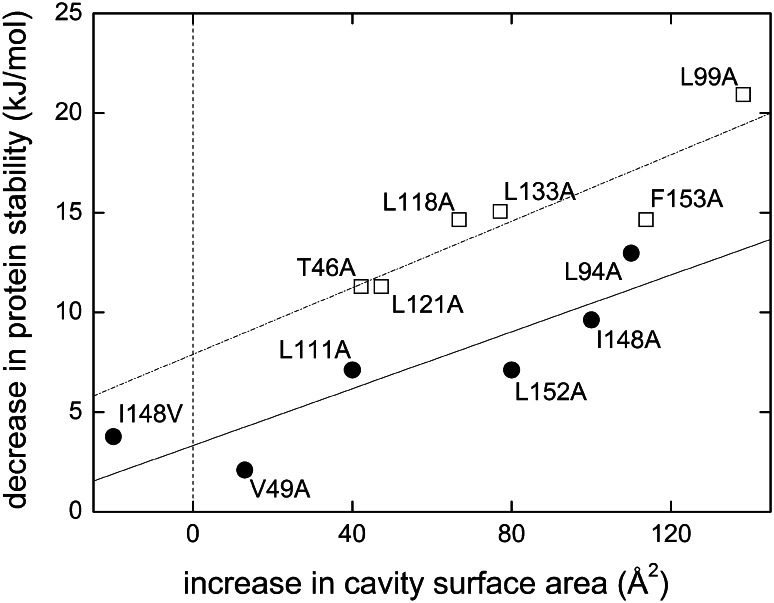
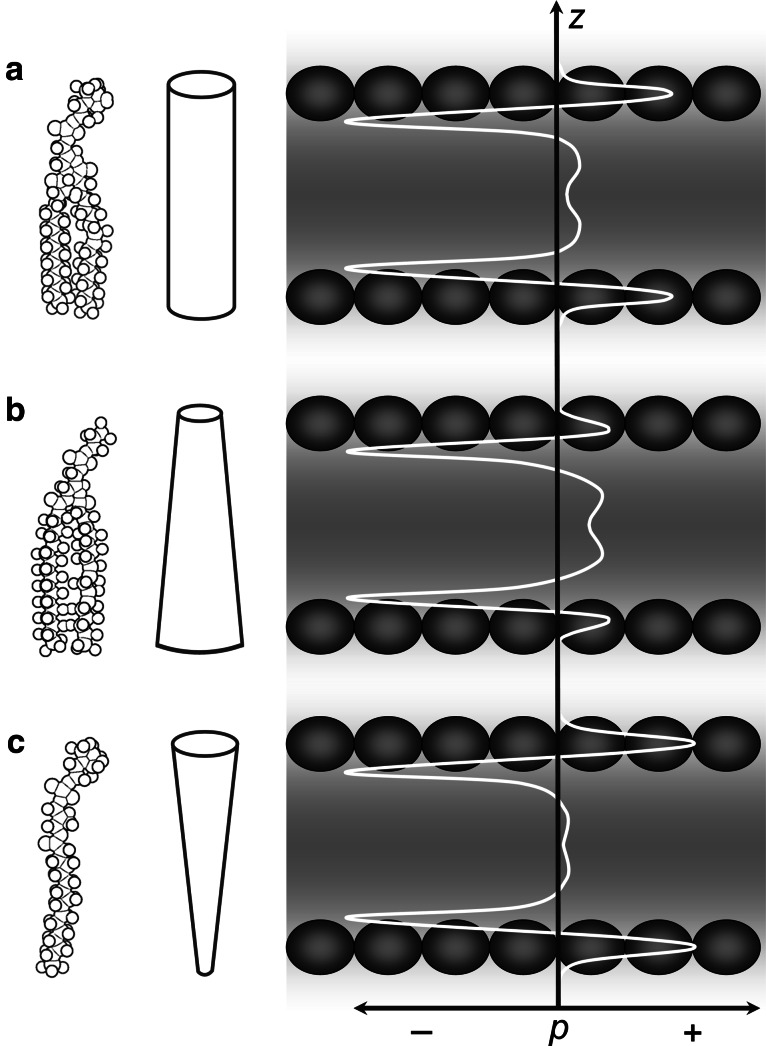
Separation of cells and organelles by bilayer membranes is a fundamental principle of life. Cellular membranes contain a baffling variety of proteins, which fulfil vital functions as receptors and signal transducers, channels and transporters, motors and anchors. The vast majority of membrane-bound proteins contain bundles of alpha-helical transmembrane domains. Understanding how these proteins adopt their native, biologically active structures in the complex milieu of a membrane is therefore a major challenge in today's life sciences. Here, we review recent progress in the folding, unfolding and refolding of alpha-helical membrane proteins and compare the molecular interactions that stabilise proteins in lipid bilayers. We also provide a critical discussion of a detergent denaturation assay that is increasingly used to determine membrane-protein stability but is not devoid of conceptual difficulties.
Figures
Similar articles
-
Lipid bilayer composition modulates the unfolding free energy of a knotted α-helical membrane protein.Proc Natl Acad Sci U S A. 2018 Feb 20;115(8):E1799-E1808. doi: 10.1073/pnas.1714668115. Epub 2018 Feb 5. Proc Natl Acad Sci U S A. 2018. PMID: 29432185 Free PMC article.
-
Membrane protein folding and stability: physical principles.Annu Rev Biophys Biomol Struct. 1999;28:319-65. doi: 10.1146/annurev.biophys.28.1.319. Annu Rev Biophys Biomol Struct. 1999. PMID: 10410805 Review.
-
Correct folding of the beta-barrel of the human membrane protein VDAC requires a lipid bilayer.J Mol Biol. 2007 Apr 20;368(1):66-78. doi: 10.1016/j.jmb.2007.01.066. Epub 2007 Feb 3. J Mol Biol. 2007. PMID: 17336328
-
Alpha-helical transmembrane peptides: a "divide and conquer" approach to membrane proteins.Chem Phys Lipids. 2010 Jan;163(1):1-26. doi: 10.1016/j.chemphyslip.2009.07.009. Chem Phys Lipids. 2010. PMID: 19682979 Review.
-
Structural insights into functional lipid-protein interactions in secondary transporters.Biochim Biophys Acta. 2015 Mar;1850(3):476-87. doi: 10.1016/j.bbagen.2014.05.010. Epub 2014 May 20. Biochim Biophys Acta. 2015. PMID: 24859688 Review.
Cited by
-
Static retention of the lumenal monotopic membrane protein torsinA in the endoplasmic reticulum.EMBO J. 2011 Jul 22;30(16):3217-31. doi: 10.1038/emboj.2011.233. EMBO J. 2011. PMID: 21785409 Free PMC article.
-
Sequential steps in the assembly of the multimeric outer membrane secretin PulD.J Biol Chem. 2013 Oct 18;288(42):30700-30707. doi: 10.1074/jbc.M113.489112. Epub 2013 Sep 9. J Biol Chem. 2013. PMID: 24019525 Free PMC article.
-
Design and characterization of a membrane protein unfolding platform in lipid bilayers.PLoS One. 2015 Mar 23;10(3):e0120253. doi: 10.1371/journal.pone.0120253. eCollection 2015. PLoS One. 2015. PMID: 25799099 Free PMC article.
-
Solid-state NMR spectroscopy based atomistic view of a membrane protein unfolding pathway.Nat Commun. 2019 Aug 27;10(1):3867. doi: 10.1038/s41467-019-11849-8. Nat Commun. 2019. PMID: 31455771 Free PMC article.
-
Genome-wide identification of the opsin protein in Leptosphaeria maculans and comparison with other fungi (pathogens of Brassica napus).Front Microbiol. 2023 Aug 25;14:1193892. doi: 10.3389/fmicb.2023.1193892. eCollection 2023. Front Microbiol. 2023. PMID: 37692395 Free PMC article.
References
-
- Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–1844. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
