

Changes in the genomic content of circulating Bordetella pertussis strains isolated from the Netherlands, Sweden, Japan and Australia: adaptive evolution or drift?
- PMID: 20102608
- PMCID: PMC2834637
- DOI: 10.1186/1471-2164-11-64
Changes in the genomic content of circulating Bordetella pertussis strains isolated from the Netherlands, Sweden, Japan and Australia: adaptive evolution or drift?
Abstract
Background: Bordetella pertussis is the causative agent of human whooping cough (pertussis) and is particularly severe in infants. Despite worldwide vaccinations, whooping cough remains a public health problem. A significant increase in the incidence of whooping cough has been observed in many countries since the 1990s. Several reasons for the re-emergence of this highly contagious disease have been suggested. A particularly intriguing possibility is based on evidence indicating that pathogen adaptation may play a role in this process. In an attempt to gain insight into the genomic make-up of B. pertussis over the last 60 years, we used an oligonucleotide DNA microarray to compare the genomic contents of a collection of 171 strains of B. pertussis isolates from different countries.
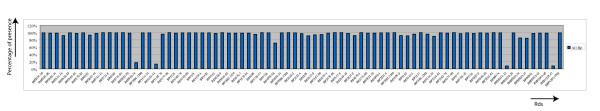
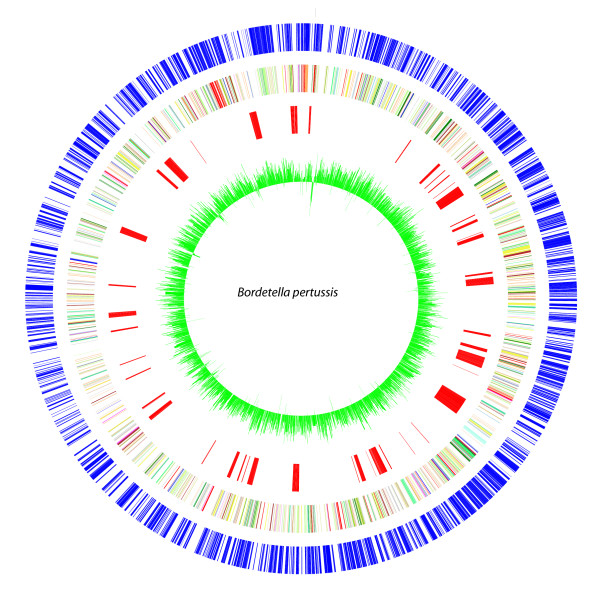
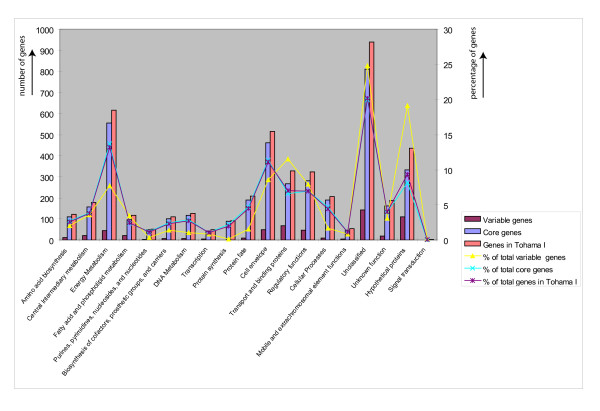
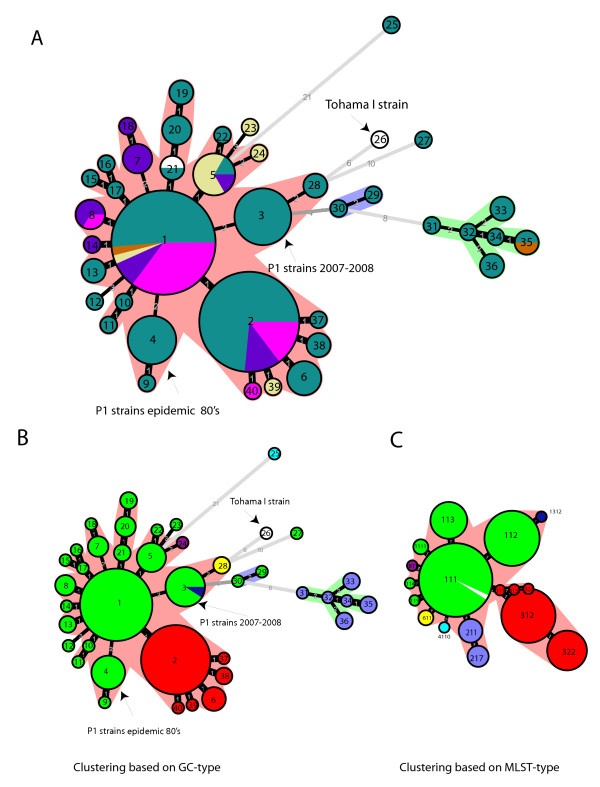
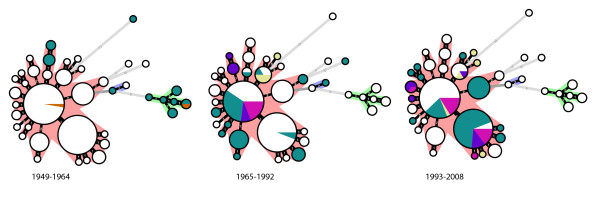
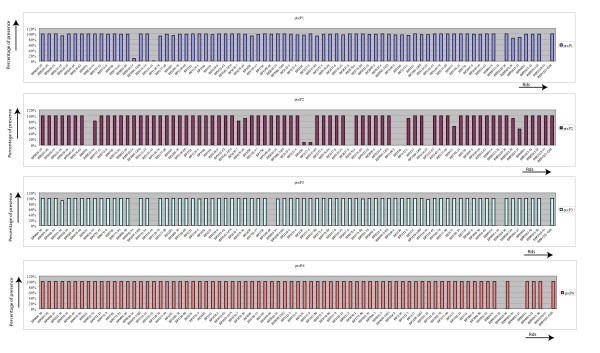
Results: The CGH microarray analysis estimated the core genome of B. pertussis, to consist of 3,281 CDSs that are conserved among all B. pertussis strains, and represent 84.8% of all CDSs found in the 171 B. pertussis strains. A total of 64 regions of difference consisting of one or more contiguous CDSs were identified among the variable genes. CGH data also revealed that the genome size of B. pertussis strains is decreasing progressively over the past 60 years. Phylogenetic analysis of microarray data generated a minimum spanning tree that depicted the phylogenetic structure of the strains. B. pertussis strains with the same gene content were found in several different countries. However, geographic specificity of the B. pertussis strains was not observed. The gene content was determined to highly correlate with the ptxP-type of the strains.
Conclusions: An overview of genomic contents of a large collection of isolates from different countries allowed us to derive a core genome and a phylogenetic structure of B. pertussis. Our results show that B. pertussis is a dynamic organism that continues to evolve.
Figures
References
-
- Heininger U, Stehr K, Schmitt-Grohe S, Lorenz C, Rost R, Christenson PD. et al. Clinical characteristics of illness caused by Bordetella parapertussis compared with illness caused by Bordetella pertussis. Pediatr Infect Dis J. 1994;13:306–309. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
