

Analysis of genotype-phenotype correlations in human holoprosencephaly
- PMID: 20104608
- PMCID: PMC2815217
- DOI: 10.1002/ajmg.c.30240
Analysis of genotype-phenotype correlations in human holoprosencephaly
Abstract
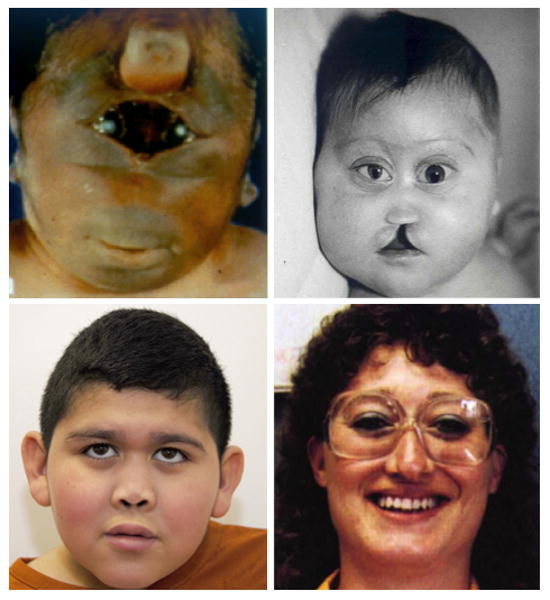
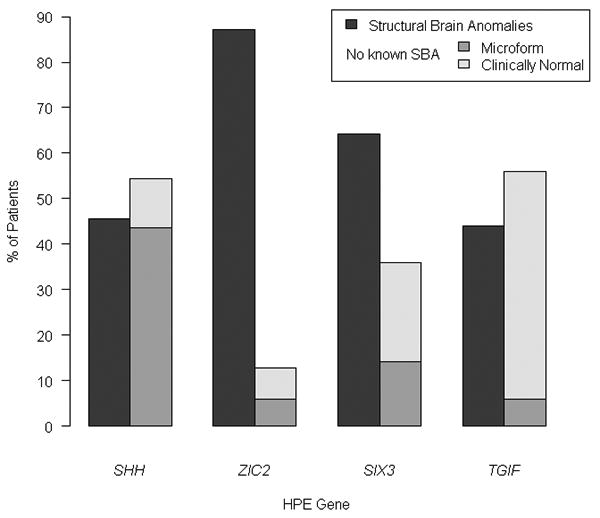
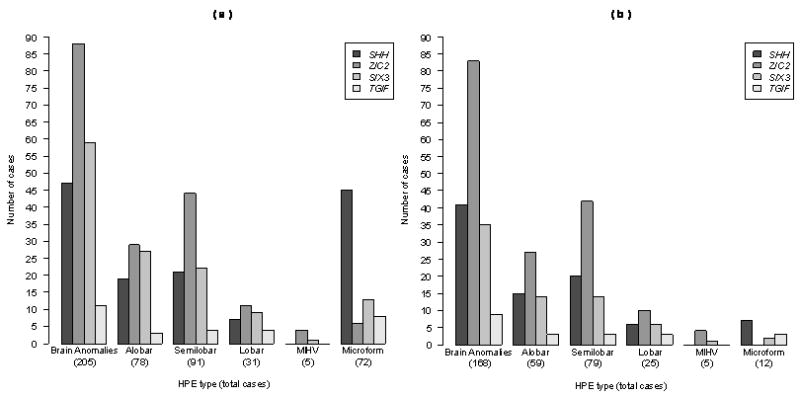
Since the discovery of the first gene causing holoprosencephaly (HPE), over 500 patients with mutations in genes associated with non-chromosomal, non-syndromic HPE have been described, with detailed descriptions available in over 300. Comprehensive clinical analysis of these individuals allows examination for the presence of genotype-phenotype correlations. These correlations allow a degree of differentiation between patients with mutations in different HPE-associated genes and for the application of functional studies to determine intragenic correlations. These early correlations are an important advance in the understanding of the clinical aspects of this disease, and in general argue for continued analysis of the genetic and clinical findings of large cohorts of patients with rare diseases in order to better inform both basic biological insight and care and counseling for affected patients and families.
2010 Wiley-Liss, Inc.
Figures
References
-
- Agresti A. An Introduction to Categorical Data Analysis. New York: John Wiley & Sons; 1996.
-
- Ardinger HH, Bartley JA. Microcephaly in familial holoprosencephaly. J Craniofac Genet Dev Biol. 1988;8:53–61. - PubMed
-
- Bendavid C, Rochard L, Dubourg C, Seguin J, Gicquel I, Pasquier L, Vigneron J, Laquerrière A, Marcorelles P, Jeanne-Pasquier C, Rouleau C, Jaillard S, Mosser J, Odent S, David V. Array-CGH analysis indicates a high prevalence of genomic rearrangements in holoprosencephaly: an updated map of candidate loci. Hum Mutat. 2009;30:1175–1182. - PubMed
-
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. - PubMed
-
- de la Cruz JM, Bamford RN, Burdine RD, Roessler E, Barkovich AJ, Donnai D, Schier AF, Muenke M. A loss-of-function mutation in the CFC domain of TDGF1 is associated with human forebrain defects. Hum Genet. 2002;110:422–428. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
