

Protease-sensitive synthetic prions
- PMID: 20107515
- PMCID: PMC2809756
- DOI: 10.1371/journal.ppat.1000736
Protease-sensitive synthetic prions
Abstract
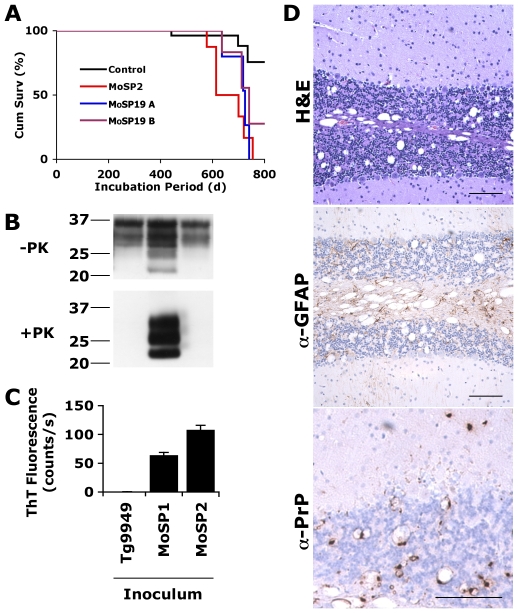
Prions arise when the cellular prion protein (PrP(C)) undergoes a self-propagating conformational change; the resulting infectious conformer is designated PrP(Sc). Frequently, PrP(Sc) is protease-resistant but protease-sensitive (s) prions have been isolated in humans and other animals. We report here that protease-sensitive, synthetic prions were generated in vitro during polymerization of recombinant (rec) PrP into amyloid fibers. In 22 independent experiments, recPrP amyloid preparations, but not recPrP monomers or oligomers, transmitted disease to transgenic mice (n = 164), denoted Tg9949 mice, that overexpress N-terminally truncated PrP. Tg9949 control mice (n = 174) did not spontaneously generate prions although they were prone to late-onset spontaneous neurological dysfunction. When synthetic prion isolates from infected Tg9949 mice were serially transmitted in the same line of mice, they exhibited sPrP(Sc) and caused neurodegeneration. Interestingly, these protease-sensitive prions did not shorten the life span of Tg9949 mice despite causing extensive neurodegeneration. We inoculated three synthetic prion isolates into Tg4053 mice that overexpress full-length PrP; Tg4053 mice are not prone to developing spontaneous neurological dysfunction. The synthetic prion isolates caused disease in 600-750 days in Tg4053 mice, which exhibited sPrP(Sc). These novel synthetic prions demonstrate that conformational changes in wild-type PrP can produce mouse prions composed exclusively of sPrP(Sc).
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


References
-
- Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SBH, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell. 1985;40:735–746. - PubMed
-
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. - PubMed
-
- McKinley MP, Bolton DC, Prusiner SB. A protease-resistant protein is a structural component of the scrapie prion. Cell. 1983;35:57–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
