

BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice
- PMID: 20107607
- PMCID: PMC2809772
- DOI: 10.1371/journal.pgen.1000826
BRIT1/MCPH1 is essential for mitotic and meiotic recombination DNA repair and maintaining genomic stability in mice
Abstract
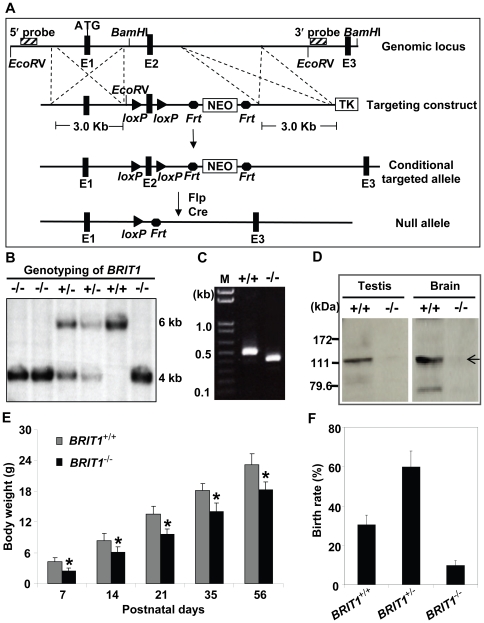
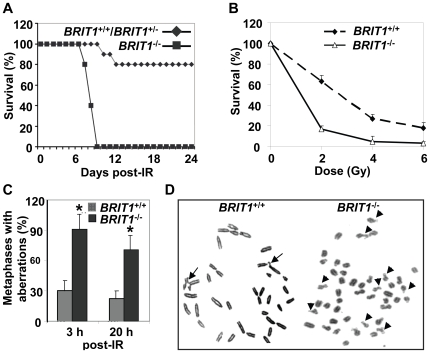
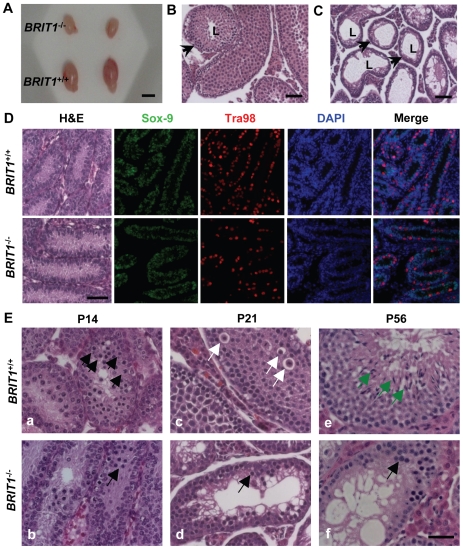
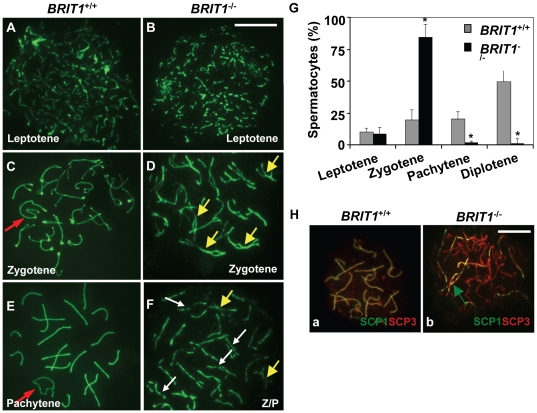
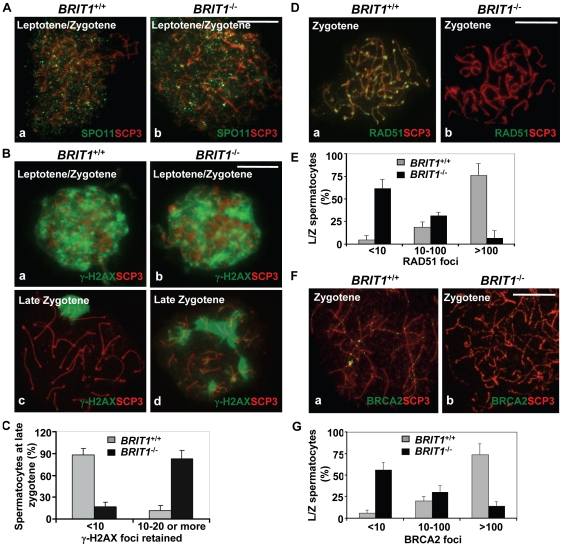
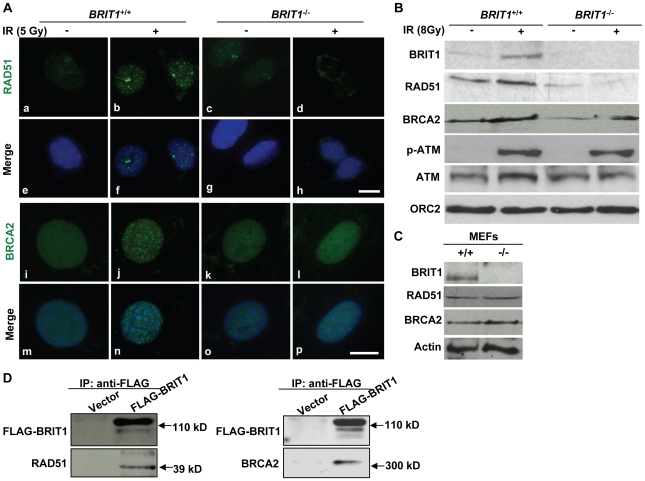
BRIT1 protein (also known as MCPH1) contains 3 BRCT domains which are conserved in BRCA1, BRCA2, and other important molecules involved in DNA damage signaling, DNA repair, and tumor suppression. BRIT1 mutations or aberrant expression are found in primary microcephaly patients as well as in cancer patients. Recent in vitro studies suggest that BRIT1/MCPH1 functions as a novel key regulator in the DNA damage response pathways. To investigate its physiological role and dissect the underlying mechanisms, we generated BRIT1(-/-) mice and identified its essential roles in mitotic and meiotic recombination DNA repair and in maintaining genomic stability. Both BRIT1(-/-) mice and mouse embryonic fibroblasts (MEFs) were hypersensitive to gamma-irradiation. BRIT1(-/-) MEFs and T lymphocytes exhibited severe chromatid breaks and reduced RAD51 foci formation after irradiation. Notably, BRIT1(-/-) mice were infertile and meiotic homologous recombination was impaired. BRIT1-deficient spermatocytes exhibited a failure of chromosomal synapsis, and meiosis was arrested at late zygotene of prophase I accompanied by apoptosis. In mutant spermatocytes, DNA double-strand breaks (DSBs) were formed, but localization of RAD51 or BRCA2 to meiotic chromosomes was severely impaired. In addition, we found that BRIT1 could bind to RAD51/BRCA2 complexes and that, in the absence of BRIT1, recruitment of RAD51 and BRCA2 to chromatin was reduced while their protein levels were not altered, indicating that BRIT1 is involved in mediating recruitment of RAD51/BRCA2 to the damage site. Collectively, our BRIT1-null mouse model demonstrates that BRIT1 is essential for maintaining genomic stability in vivo to protect the hosts from both programmed and irradiation-induced DNA damages, and its depletion causes a failure in both mitotic and meiotic recombination DNA repair via impairing RAD51/BRCA2's function and as a result leads to infertility and genomic instability in mice.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Multiple roles of BRIT1/MCPH1 in DNA damage response, DNA repair, and cancer suppression.Yonsei Med J. 2010 May;51(3):295-301. doi: 10.3349/ymj.2010.51.3.295. Yonsei Med J. 2010. PMID: 20376879 Free PMC article. Review.
-
Mcph1/Brit1 deficiency promotes genomic instability and tumor formation in a mouse model.Oncogene. 2015 Aug 13;34(33):4368-78. doi: 10.1038/onc.2014.367. Epub 2014 Nov 3. Oncogene. 2015. PMID: 25362854 Free PMC article.
-
DNA damage response in microcephaly development of MCPH1 mouse model.DNA Repair (Amst). 2013 Aug;12(8):645-55. doi: 10.1016/j.dnarep.2013.04.017. Epub 2013 May 15. DNA Repair (Amst). 2013. PMID: 23683352
-
A meiosis-specific BRCA2 binding protein recruits recombinases to DNA double-strand breaks to ensure homologous recombination.Nat Commun. 2019 Feb 13;10(1):722. doi: 10.1038/s41467-019-08676-2. Nat Commun. 2019. PMID: 30760716 Free PMC article.
-
Biochemical attributes of mitotic and meiotic presynaptic complexes.DNA Repair (Amst). 2018 Nov;71:148-157. doi: 10.1016/j.dnarep.2018.08.018. Epub 2018 Aug 23. DNA Repair (Amst). 2018. PMID: 30195641 Free PMC article. Review.
Cited by
-
Essential developmental, genomic stability, and tumour suppressor functions of the mouse orthologue of hSSB1/NABP2.PLoS Genet. 2013;9(2):e1003298. doi: 10.1371/journal.pgen.1003298. Epub 2013 Feb 7. PLoS Genet. 2013. PMID: 23408915 Free PMC article.
-
Double-strand break repair on sex chromosomes: challenges during male meiotic prophase.Cell Cycle. 2015;14(4):516-25. doi: 10.1080/15384101.2014.998070. Cell Cycle. 2015. PMID: 25565522 Free PMC article. Review.
-
Analysis of the "centrosome-ome" identifies MCPH1 deletion as a cause of centrosome amplification in human cancer.Sci Rep. 2020 Jul 17;10(1):11921. doi: 10.1038/s41598-020-68629-4. Sci Rep. 2020. PMID: 32681070 Free PMC article.
-
MCPH1, beyond its role deciding the brain size.Aging (Albany NY). 2021 Oct 27;13(20):23437-23439. doi: 10.18632/aging.203658. Epub 2021 Oct 27. Aging (Albany NY). 2021. PMID: 34705666 Free PMC article. No abstract available.
-
Neurogenesis in primates versus rodents and the value of non-human primate models.Natl Sci Rev. 2023 Sep 15;10(11):nwad248. doi: 10.1093/nsr/nwad248. eCollection 2023 Nov. Natl Sci Rev. 2023. PMID: 38025664 Free PMC article. Review.
References
-
- Pastink A, Eeken JC, Lohman PH. Genomic integrity and the repair of double-strand DNA breaks. Mutat Res. 2001;480–481:37–50. - PubMed
-
- Franco S, Alt FW, Manis JP. Pathways that suppress programmed DNA breaks from progressing to chromosomal breaks and translocations. DNA Repair. 2006;5:1030–1041. - PubMed
-
- Pandita TK, Hittelman WN. The contribution of DNA and chromosome repair deficiencies to the radiosensitivity of ataxia-telangiectasia. Radiat Res. 1992;131:214–223. - PubMed
-
- Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. - PubMed
-
- Richardson C, Horikoshi N, Pandita TK. The role of the DNA double-strand break response network in meiosis. DNA Repair. 2004;3:1149–1164. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
