

Evolutionary trajectories of beta-lactamase CTX-M-1 cluster enzymes: predicting antibiotic resistance
- PMID: 20107608
- PMCID: PMC2809773
- DOI: 10.1371/journal.ppat.1000735
Evolutionary trajectories of beta-lactamase CTX-M-1 cluster enzymes: predicting antibiotic resistance
Abstract
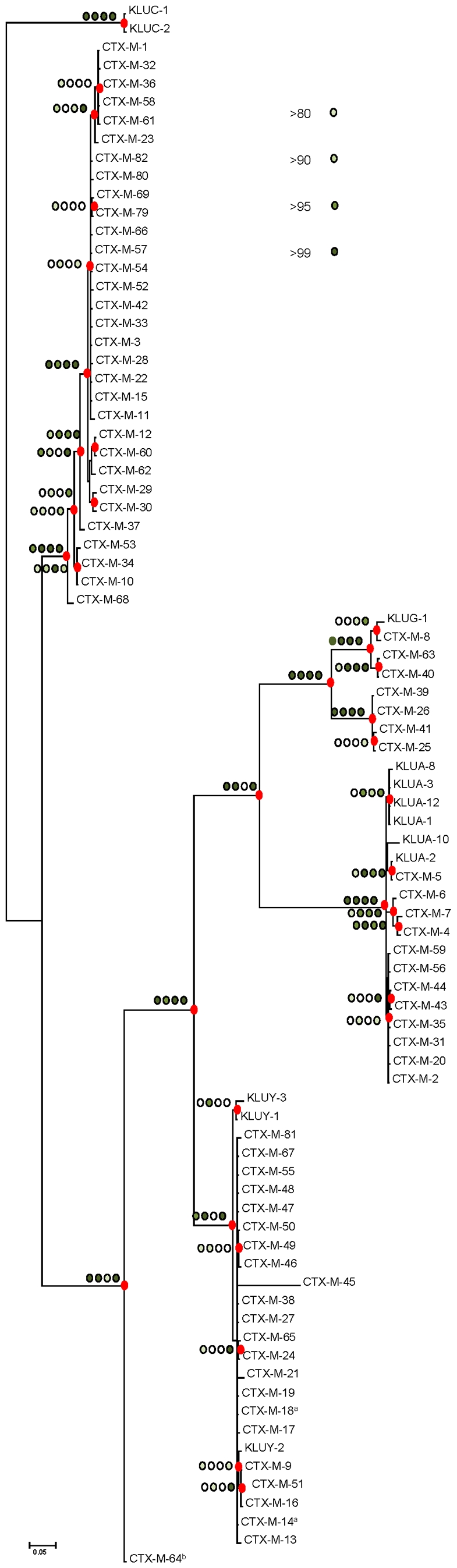
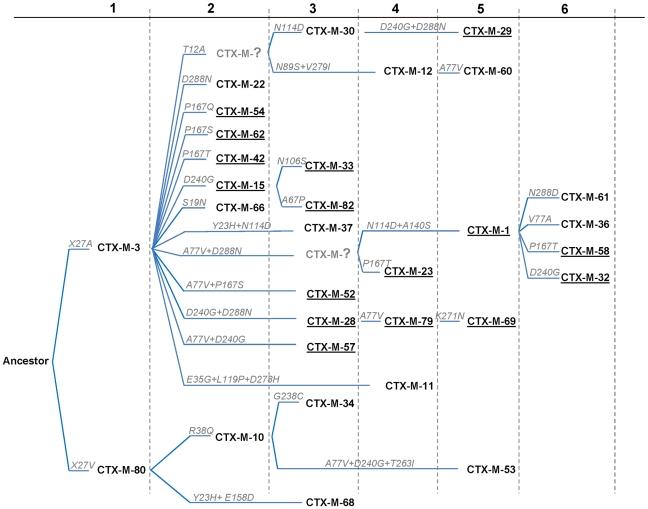
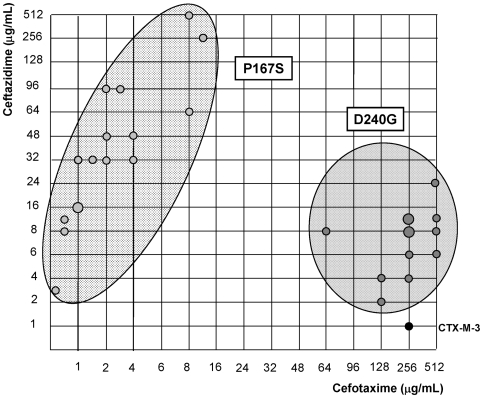
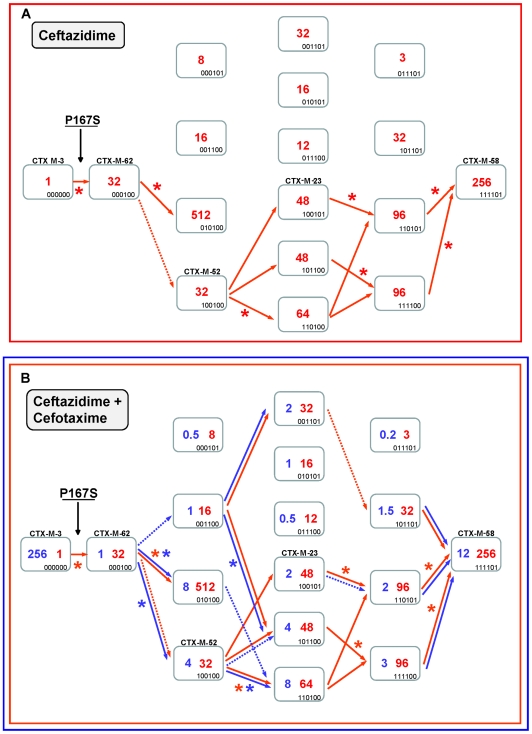
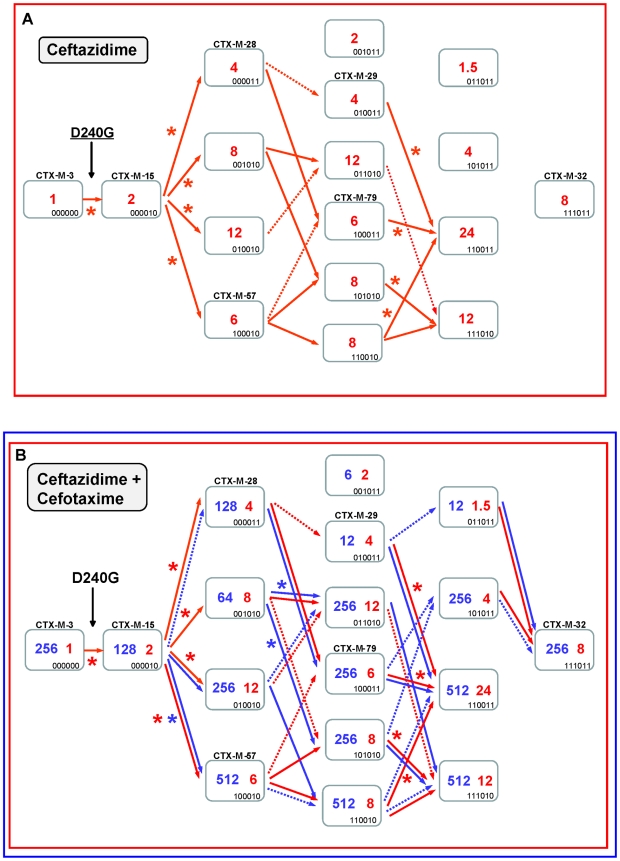
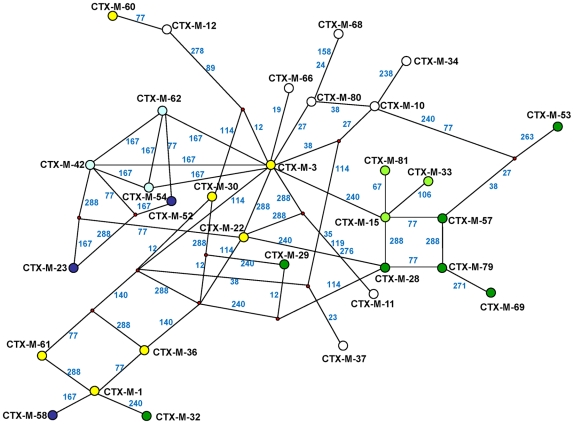
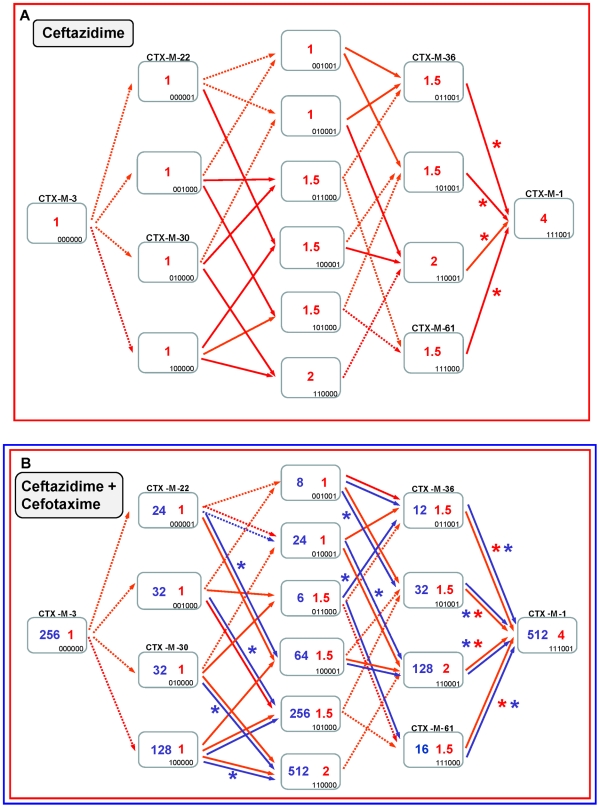
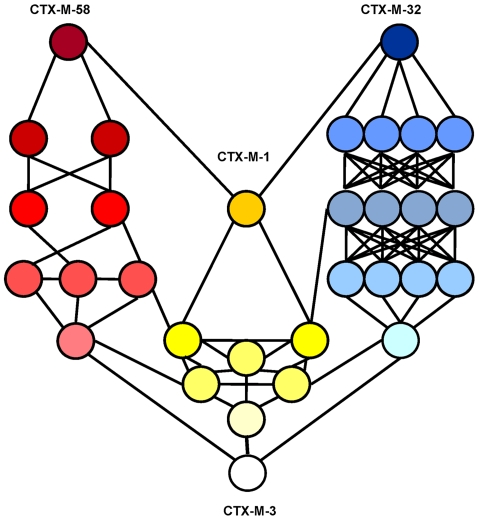
Extended-spectrum beta-lactamases (ESBL) constitute a key antibiotic-resistance mechanism affecting Gram-negative bacteria, and also an excellent model for studying evolution in real time. A shift in the epidemiology of ESBLs is being observed, which is characterized by the explosive diversification and increase in frequency of the CTX-M-type beta-lactamases in different settings. This provides a unique opportunity for studying a protein evolutionary radiation by the sequential acquisition of specific mutations enhancing protein efficiency and fitness concomitantly. The existence of driver antibiotic molecules favoring protein divergence has been investigated by combining evolutionary analyses and experimental site-specific mutagenesis. Phylogenetic reconstruction with all the CTX-M variants described so far provided a hypothetical evolutionary scenario showing at least three diversification events. CTX-M-3 was likely the enzyme at the origin of the diversification in the CTX-M-1 cluster, which was coincident with positive selection acting on several amino acid positions. Sixty-three CTX-M-3 derivatives containing all combinations of mutations under positively selected positions were constructed, and their phenotypic efficiency was evaluated. The CTX-M-3 diversification process can only be explained in a complex selective landscape with at least two antibiotics (cefotaxime and ceftazidime), indicating the need to invoke mixtures of selective drivers in order to understand the final evolutionary outcome. Under this hypothesis, we found congruent results between the in silico and in vitro analyses of evolutionary trajectories. Three pathways driving the diversification of CTX-M-3 towards the most complex and efficient variants were identified. Whereas the P167S pathway has limited possibilities of further diversification, the D240G route shows a robust diversification network. In the third route, drift may have played a role in the early stages of CTX-M-3 evolution. Antimicrobial agents should not be considered only as selectors for efficient mechanisms of resistance but also as diversifying agents of the evolutionary trajectories. Different trajectories were identified using a combination of phylogenetic reconstructions and directed mutagenesis analyses, indicating that such an approach might be useful to fulfill the desirable goal of predicting evolutionary trajectories in antimicrobial resistance.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Canton R, Coque TM. The CTX-M beta-lactamase pandemic. Curr Opin Microbiol. 2006;9:466–475. - PubMed
-
- Pitout JD, Laupland KB. Extended-spectrum beta-lactamase-producing Enterobacteriaceae: an emerging public-health concern. Lancet Infect Dis. 2008;8:159–166. - PubMed
-
- Petrosino J, Cantu C, III, Palzkill T. Beta-Lactamases: protein evolution in real time. Trends Microbiol. 1998;6:323–327. - PubMed
-
- Gniadkowski M. Evolution of extended-spectrum beta-lactamases by mutation. Clin Microbiol Infect. 2008;14(Suppl 1):11–32. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
