

Integrating the multiple dimensions of genomic and epigenomic landscapes of cancer
- PMID: 20108112
- PMCID: PMC3415277
- DOI: 10.1007/s10555-010-9199-2
Integrating the multiple dimensions of genomic and epigenomic landscapes of cancer
Abstract
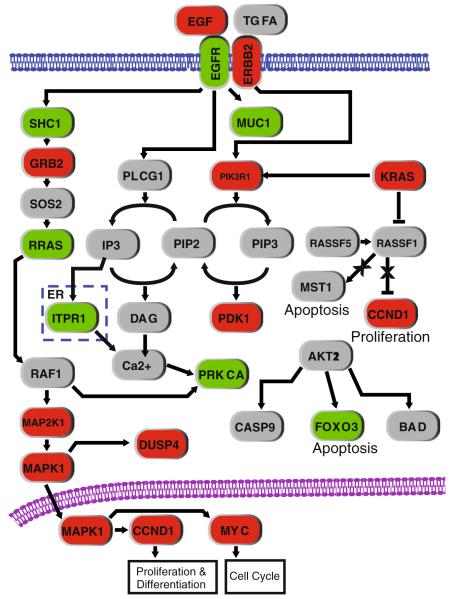
Advances in high-throughput, genome-wide profiling technologies have allowed for an unprecedented view of the cancer genome landscape. Specifically, high-density microarrays and sequencing-based strategies have been widely utilized to identify genetic (such as gene dosage, allelic status, and mutations in gene sequence) and epigenetic (such as DNA methylation, histone modification, and microRNA) aberrations in cancer. Although the application of these profiling technologies in unidimensional analyses has been instrumental in cancer gene discovery, genes affected by low-frequency events are often overlooked. The integrative approach of analyzing parallel dimensions has enabled the identification of (a) genes that are often disrupted by multiple mechanisms but at low frequencies by any one mechanism and (b) pathways that are often disrupted at multiple components but at low frequencies at individual components. These benefits of using an integrative approach illustrate the concept that the whole is greater than the sum of its parts. As efforts have now turned toward parallel and integrative multidimensional approaches for studying the cancer genome landscape in hopes of obtaining a more insightful understanding of the key genes and pathways driving cancer cells, this review describes key findings disseminating from such high-throughput, integrative analyses, including contributions to our understanding of causative genetic events in cancer cell biology.
Figures

Similar articles
-
An integrative multi-dimensional genetic and epigenetic strategy to identify aberrant genes and pathways in cancer.BMC Syst Biol. 2010 May 17;4:67. doi: 10.1186/1752-0509-4-67. BMC Syst Biol. 2010. PMID: 20478067 Free PMC article.
-
SIGMA2: a system for the integrative genomic multi-dimensional analysis of cancer genomes, epigenomes, and transcriptomes.BMC Bioinformatics. 2008 Oct 7;9:422. doi: 10.1186/1471-2105-9-422. BMC Bioinformatics. 2008. PMID: 18840289 Free PMC article.
-
Integration of genome scale data for identifying new players in colorectal cancer.World J Gastroenterol. 2016 Jan 14;22(2):534-45. doi: 10.3748/wjg.v22.i2.534. World J Gastroenterol. 2016. PMID: 26811605 Free PMC article. Review.
-
Whole genome profiling and other high throughput technologies in lymphoid neoplasms--current contributions and future hopes.Mod Pathol. 2013 Jan;26 Suppl 1:S97-S110. doi: 10.1038/modpathol.2012.179. Mod Pathol. 2013. PMID: 23281439 Review.
-
Highly parallel genomic assays.Nat Rev Genet. 2006 Aug;7(8):632-44. doi: 10.1038/nrg1901. Nat Rev Genet. 2006. PMID: 16847463 Review.
Cited by
-
Identification of tumor suppressors and oncogenes from genomic and epigenetic features in ovarian cancer.PLoS One. 2011;6(12):e28503. doi: 10.1371/journal.pone.0028503. Epub 2011 Dec 8. PLoS One. 2011. PMID: 22174824 Free PMC article.
-
YEATS4 is a novel oncogene amplified in non-small cell lung cancer that regulates the p53 pathway.Cancer Res. 2013 Dec 15;73(24):7301-12. doi: 10.1158/0008-5472.CAN-13-1897. Epub 2013 Oct 29. Cancer Res. 2013. PMID: 24170126 Free PMC article.
-
DNA methylation analysis in plants: review of computational tools and future perspectives.Brief Bioinform. 2020 May 21;21(3):906-918. doi: 10.1093/bib/bbz039. Brief Bioinform. 2020. PMID: 31220217 Free PMC article. Review.
-
Integrative miRNA and mRNA analysis in penile carcinomas reveals markers and pathways with potential clinical impact.Oncotarget. 2017 Feb 28;8(9):15294-15306. doi: 10.18632/oncotarget.14783. Oncotarget. 2017. PMID: 28122331 Free PMC article.
-
Strategies for Integrated Analysis of Genetic, Epigenetic, and Gene Expression Variation in Cancer: Addressing the Challenges.Front Genet. 2016 Feb 1;7:2. doi: 10.3389/fgene.2016.00002. eCollection 2016. Front Genet. 2016. PMID: 26870081 Free PMC article. Review.
References
-
- Pinkel D, Segraves R, Sudar D, Clark S, Poole I, Kowbel D, et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature Genetics. 1998;20(2):207–211. - PubMed
-
- Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson-Smith MA, et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273(5274):494–497. - PubMed
-
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2009;327(5961):78–81. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
