

Building and refining protein models within cryo-electron microscopy density maps based on homology modeling and multiscale structure refinement
- PMID: 20109465
- PMCID: PMC2860449
- DOI: 10.1016/j.jmb.2010.01.041
Building and refining protein models within cryo-electron microscopy density maps based on homology modeling and multiscale structure refinement
Abstract
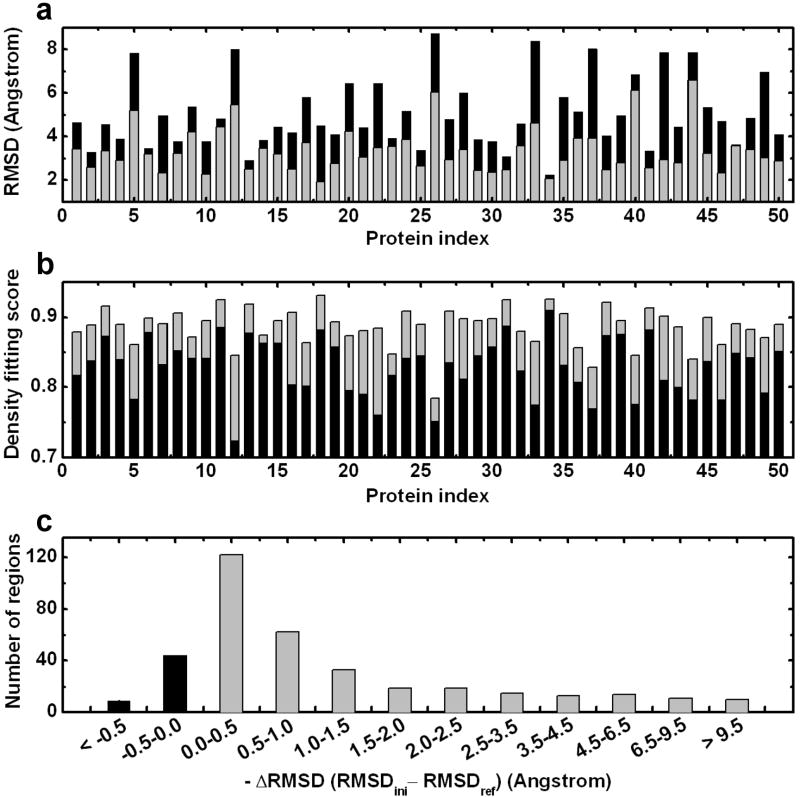
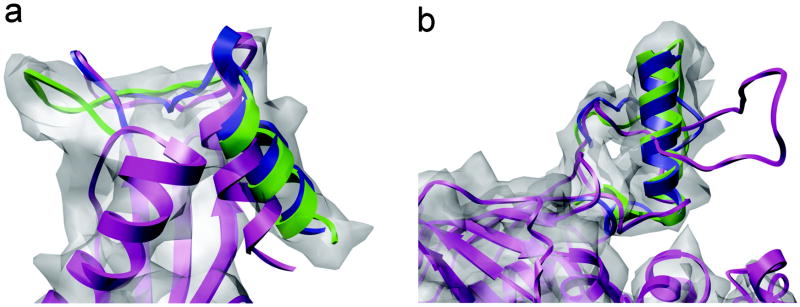
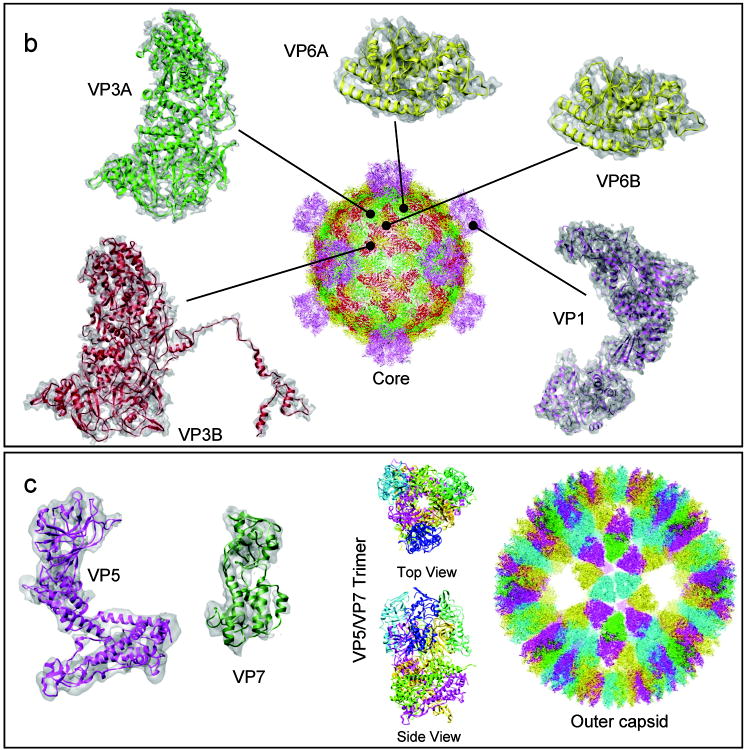
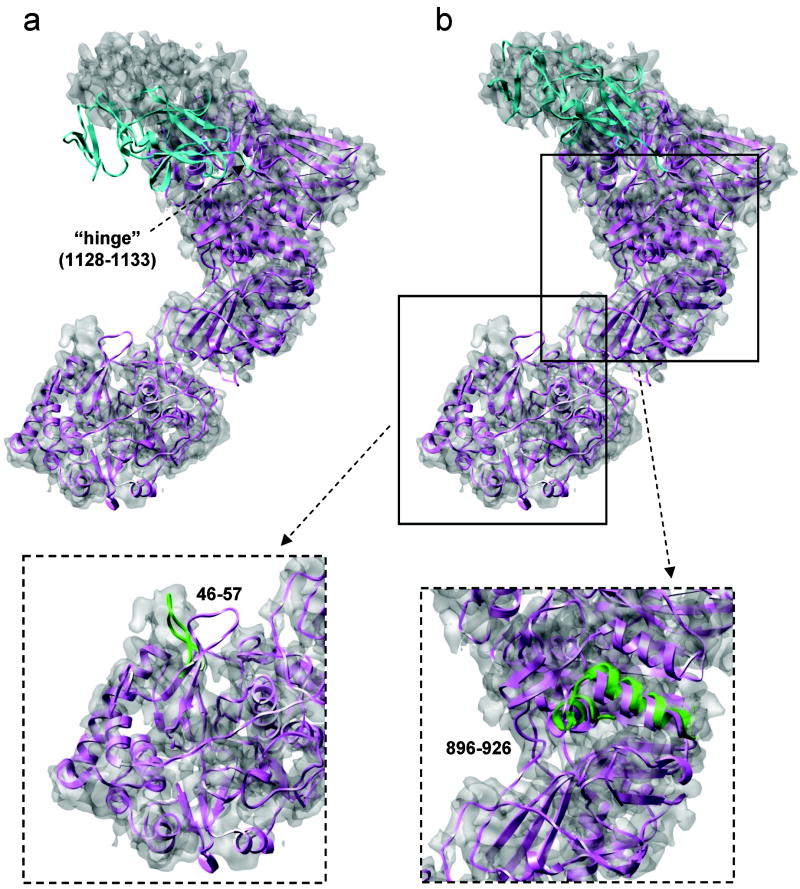
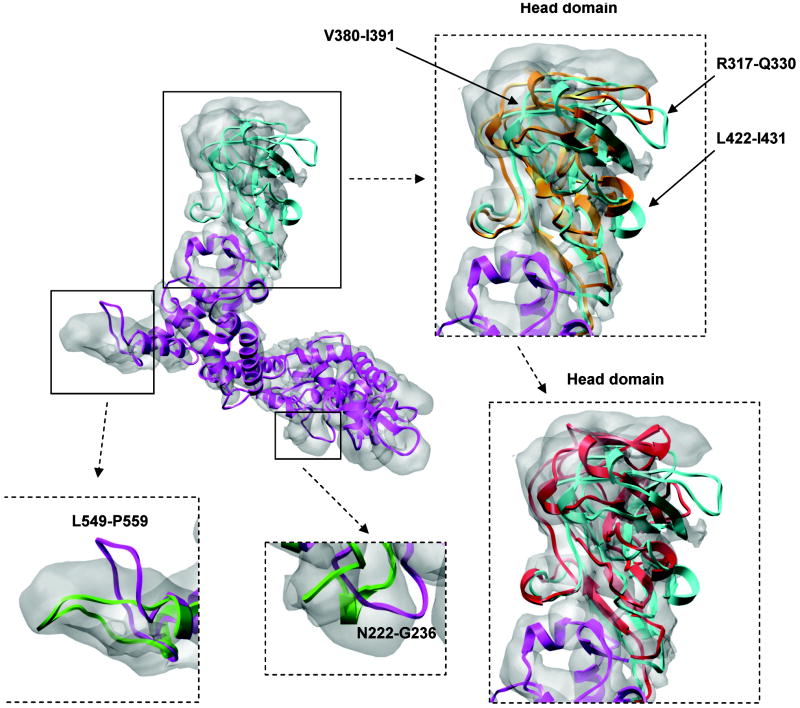
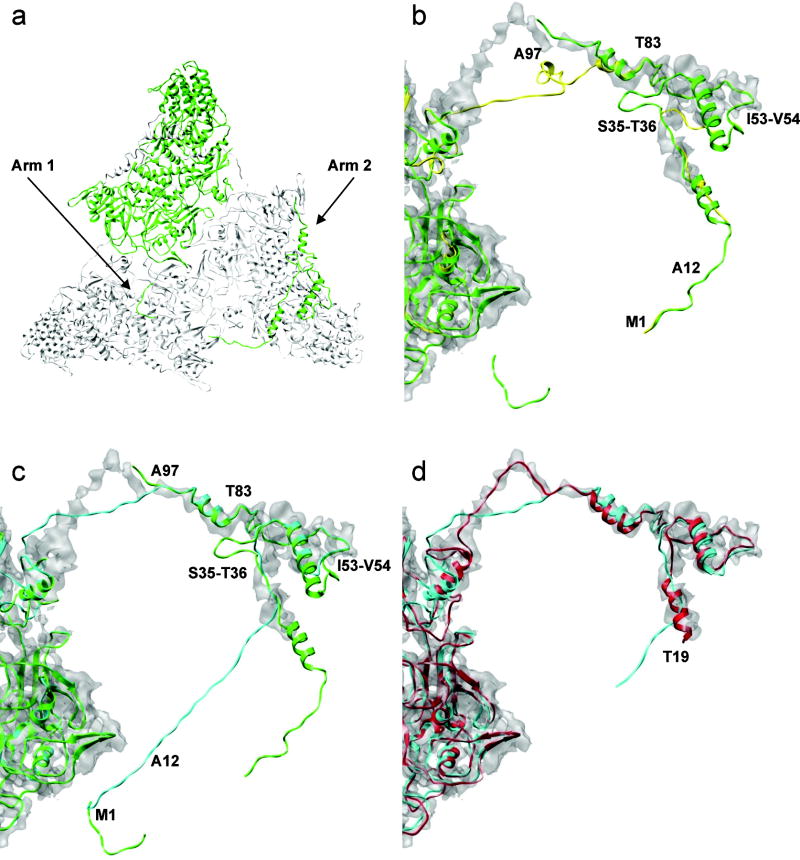
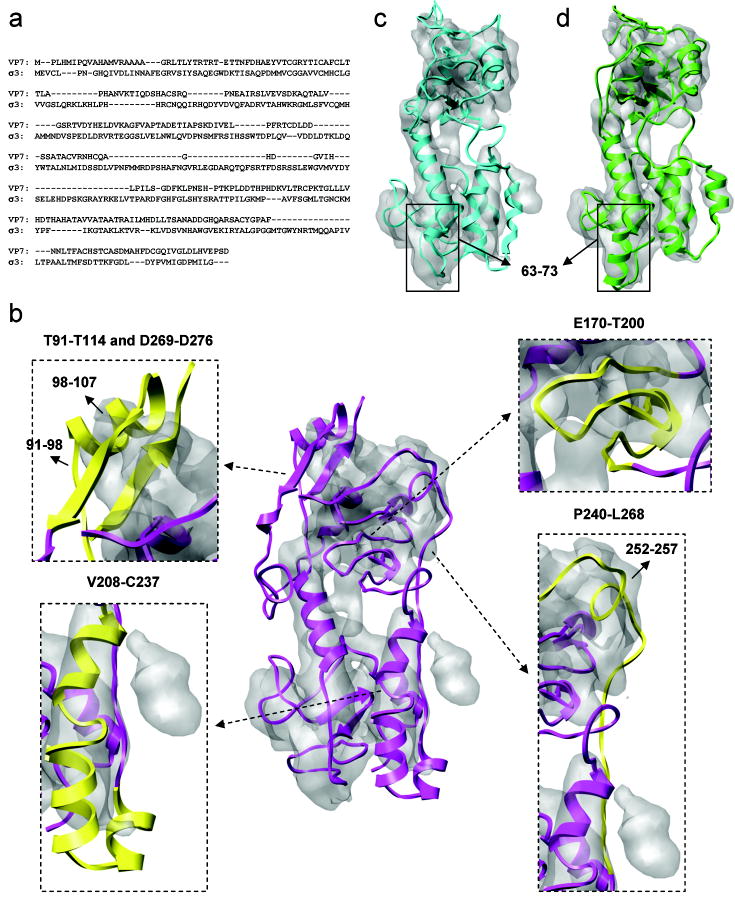
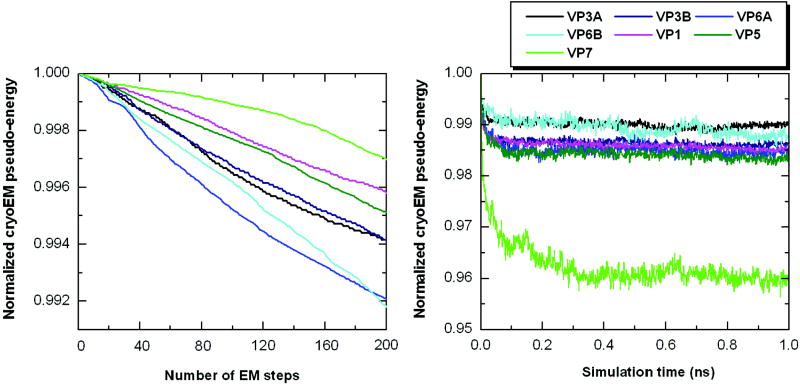
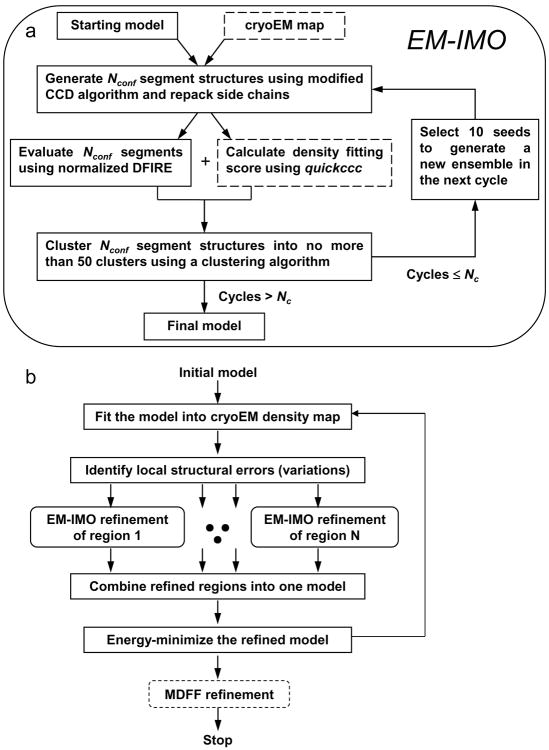
Automatic modeling methods using cryoelectron microscopy (cryoEM) density maps as constraints are promising approaches to building atomic models of individual proteins or protein domains. However, their application to large macromolecular assemblies has not been possible largely due to computational limitations inherent to such unsupervised methods. Here we describe a new method, EM-IMO (electron microscopy-iterative modular optimization), for building, modifying and refining local structures of protein models using cryoEM maps as a constraint. As a supervised refinement method, EM-IMO allows users to specify parameters derived from inspections so as to guide, and as a consequence, significantly speed up the refinement. An EM-IMO-based refinement protocol is first benchmarked on a data set of 50 homology models using simulated density maps. A multiscale refinement strategy that combines EM-IMO-based and molecular dynamics-based refinement is then applied to build backbone models for the seven conformers of the five capsid proteins in our near-atomic-resolution cryoEM map of the grass carp reovirus virion, a member of the Aquareovirus genus of the Reoviridae family. The refined models allow us to reconstruct a backbone model of the entire grass carp reovirus capsid and provide valuable functional insights that are described in the accompanying publication [Cheng, L., Zhu, J., Hui, W. H., Zhang, X., Honig, B., Fang, Q. & Zhou, Z. H. (2010). Backbone model of an aquareovirus virion by cryo-electron microscopy and bioinformatics. J. Mol. Biol. (this issue). doi:10.1016/j.jmb.2009.12.027.]. Our study demonstrates that the integrated use of homology modeling and a multiscale refinement protocol that combines supervised and automated structure refinement offers a practical strategy for building atomic models based on medium- to high-resolution cryoEM density maps.
Copyright 2010 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Backbone model of an aquareovirus virion by cryo-electron microscopy and bioinformatics.J Mol Biol. 2010 Apr 2;397(3):852-63. doi: 10.1016/j.jmb.2009.12.027. Epub 2009 Dec 28. J Mol Biol. 2010. PMID: 20036256 Free PMC article.
-
Using NMR Chemical Shifts and Cryo-EM Density Restraints in Iterative Rosetta-MD Protein Structure Refinement.J Chem Inf Model. 2020 May 26;60(5):2522-2532. doi: 10.1021/acs.jcim.9b00932. Epub 2019 Dec 24. J Chem Inf Model. 2020. PMID: 31872764 Free PMC article.
-
A New Protocol for Atomic-Level Protein Structure Modeling and Refinement Using Low-to-Medium Resolution Cryo-EM Density Maps.J Mol Biol. 2020 Sep 4;432(19):5365-5377. doi: 10.1016/j.jmb.2020.07.027. Epub 2020 Aug 6. J Mol Biol. 2020. PMID: 32771523 Free PMC article.
-
Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM.Acta Crystallogr D Struct Biol. 2018 Jun 1;74(Pt 6):492-505. doi: 10.1107/S2059798318007313. Epub 2018 May 30. Acta Crystallogr D Struct Biol. 2018. PMID: 29872001 Free PMC article. Review.
-
Tools for Model Building and Optimization into Near-Atomic Resolution Electron Cryo-Microscopy Density Maps.Methods Enzymol. 2016;579:255-76. doi: 10.1016/bs.mie.2016.06.003. Epub 2016 Aug 12. Methods Enzymol. 2016. PMID: 27572730 Free PMC article. Review.
Cited by
-
Advances in Structure Modeling Methods for Cryo-Electron Microscopy Maps.Molecules. 2019 Dec 24;25(1):82. doi: 10.3390/molecules25010082. Molecules. 2019. PMID: 31878333 Free PMC article. Review.
-
Homology modeling in the time of collective and artificial intelligence.Comput Struct Biotechnol J. 2020 Nov 14;18:3494-3506. doi: 10.1016/j.csbj.2020.11.007. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 33304450 Free PMC article. Review.
-
Exploring the spatial and temporal organization of a cell's proteome.J Struct Biol. 2011 Mar;173(3):483-96. doi: 10.1016/j.jsb.2010.11.011. Epub 2010 Nov 19. J Struct Biol. 2011. PMID: 21094684 Free PMC article. Review.
-
Automated protein structure modeling with SWISS-MODEL Workspace and the Protein Model Portal.Methods Mol Biol. 2012;857:107-36. doi: 10.1007/978-1-61779-588-6_5. Methods Mol Biol. 2012. PMID: 22323219 Free PMC article.
-
Crystal structure of PG16 and chimeric dissection with somatically related PG9: structure-function analysis of two quaternary-specific antibodies that effectively neutralize HIV-1.J Virol. 2010 Aug;84(16):8098-110. doi: 10.1128/JVI.00966-10. Epub 2010 Jun 10. J Virol. 2010. PMID: 20538861 Free PMC article.
References
-
- Chiu W, Baker ML, Almo SC. Structural biology of cellular machines. Trends in Cell Biology. 2006;16:144–150. - PubMed
-
- Mitra K, Frank J. Ribosome dynamics: Insights from atomic structure modeling into cryo-electron microscopy maps. Annual Review of Biophysics and Biomolecular Structure. 2006;35:299–317. - PubMed
-
- Jiang W, et al. Backbone structure of the infectious epsilon 15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–U12. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
