

Splicing factor and exon profiling across human tissues
- PMID: 20110256
- PMCID: PMC2875023
- DOI: 10.1093/nar/gkq008
Splicing factor and exon profiling across human tissues
Abstract
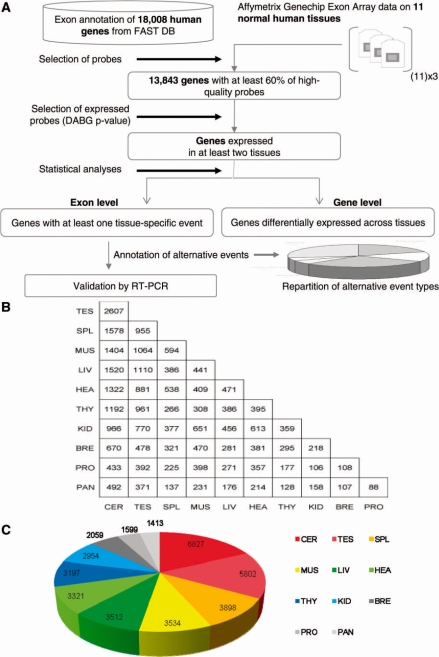
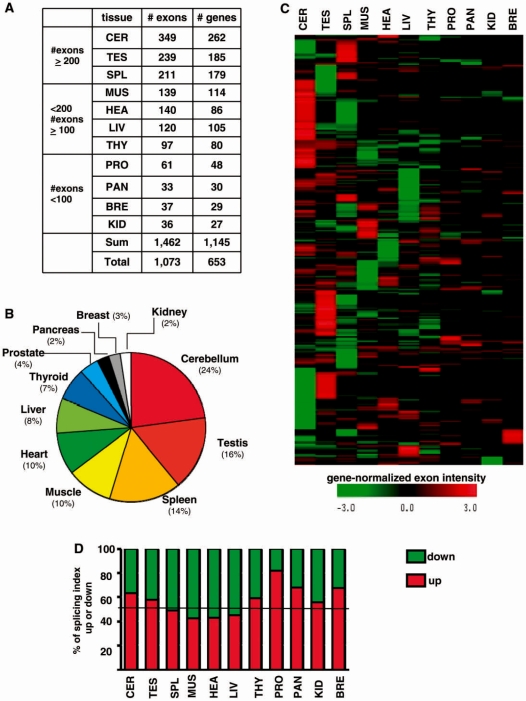
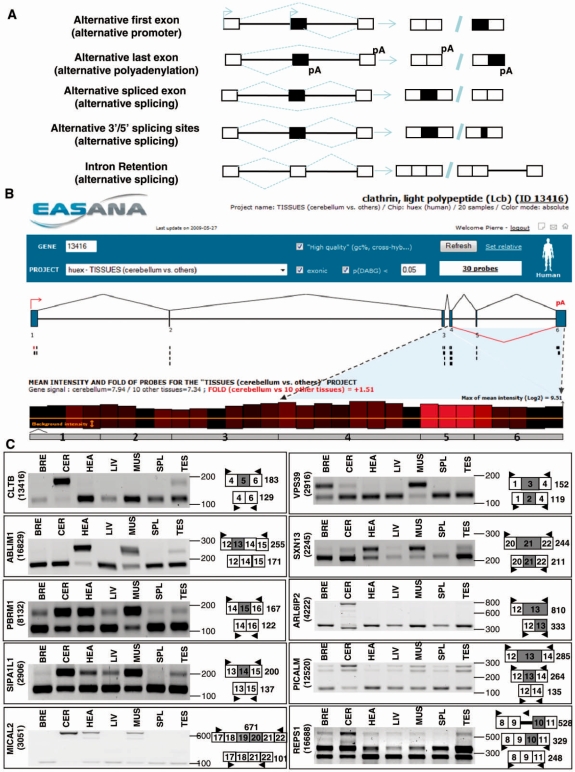
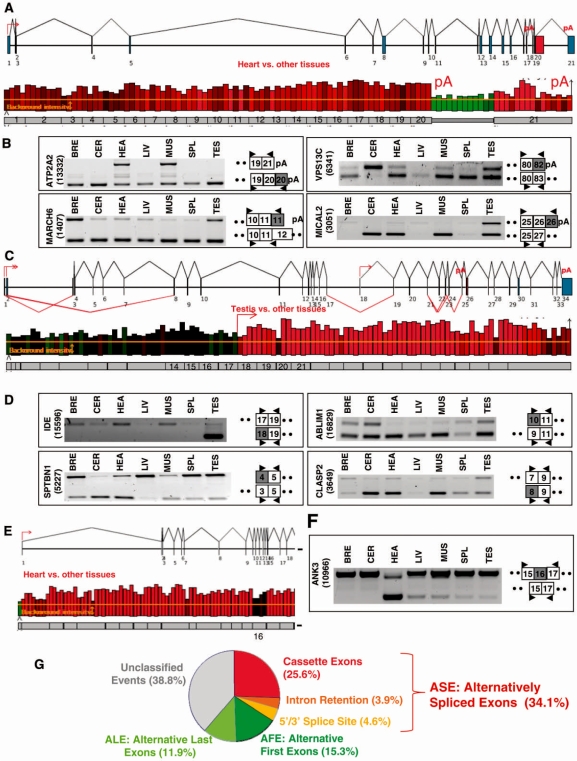
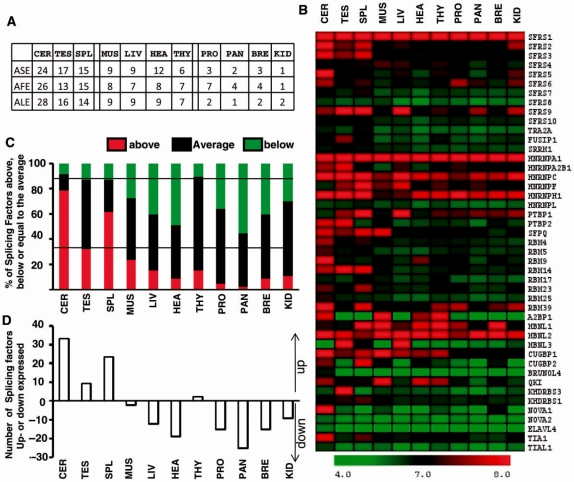
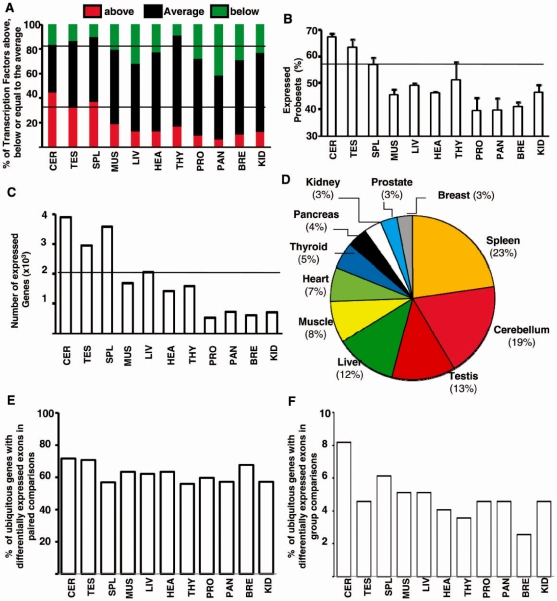
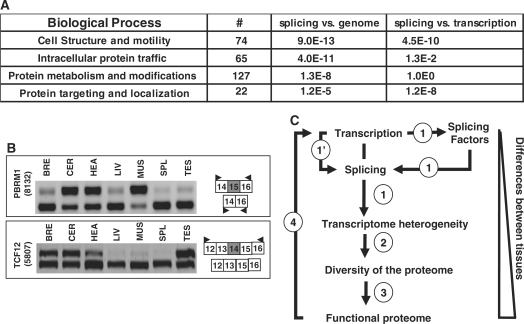
It has been shown that alternative splicing is especially prevalent in brain and testis when compared to other tissues. To test whether there is a specific propensity of these tissues to generate splicing variants, we used a single source of high-density microarray data to perform both splicing factor and exon expression profiling across 11 normal human tissues. Paired comparisons between tissues and an original exon-based statistical group analysis demonstrated after extensive RT-PCR validation that the cerebellum, testis, and spleen had the largest proportion of differentially expressed alternative exons. Variations at the exon level correlated with a larger number of splicing factors being expressed at a high level in the cerebellum, testis and spleen than in other tissues. However, this splicing factor expression profile was similar to a more global gene expression pattern as a larger number of genes had a high expression level in the cerebellum, testis and spleen. In addition to providing a unique resource on expression profiling of alternative splicing variants and splicing factors across human tissues, this study demonstrates that the higher prevalence of alternative splicing in a subset of tissues originates from the larger number of genes, including splicing factors, being expressed than in other tissues.
Figures
Similar articles
-
Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20.Clin Cancer Res. 2004 Jul 15;10(14):4652-60. doi: 10.1158/1078-0432.CCR-03-0439. Clin Cancer Res. 2004. PMID: 15269137
-
Discovery of tissue-specific exons using comprehensive human exon microarrays.Genome Biol. 2007;8(4):R64. doi: 10.1186/gb-2007-8-4-r64. Genome Biol. 2007. PMID: 17456239 Free PMC article.
-
Alternative splicing and differential gene expression in colon cancer detected by a whole genome exon array.BMC Genomics. 2006 Dec 27;7:325. doi: 10.1186/1471-2164-7-325. BMC Genomics. 2006. PMID: 17192196 Free PMC article.
-
Endothelial, epithelial, and fibroblast cells exhibit specific splicing programs independently of their tissue of origin.Genome Res. 2014 Mar;24(3):511-21. doi: 10.1101/gr.162933.113. Epub 2013 Dec 4. Genome Res. 2014. PMID: 24307554 Free PMC article.
-
Tissue-specific splicing factor gene expression signatures.Nucleic Acids Res. 2008 Sep;36(15):4823-32. doi: 10.1093/nar/gkn463. Epub 2008 Jul 24. Nucleic Acids Res. 2008. PMID: 18653532 Free PMC article.
Cited by
-
Mutation of a U2 snRNA gene causes global disruption of alternative splicing and neurodegeneration.Cell. 2012 Jan 20;148(1-2):296-308. doi: 10.1016/j.cell.2011.11.057. Cell. 2012. PMID: 22265417 Free PMC article.
-
Characterising the RNA-binding protein atlas of the mammalian brain uncovers RBM5 misregulation in mouse models of Huntington's disease.Nat Commun. 2023 Jul 19;14(1):4348. doi: 10.1038/s41467-023-39936-x. Nat Commun. 2023. PMID: 37468457 Free PMC article.
-
Aberrant splicing prediction across human tissues.Nat Genet. 2023 May;55(5):861-870. doi: 10.1038/s41588-023-01373-3. Epub 2023 May 4. Nat Genet. 2023. PMID: 37142848
-
RNA-Seq of Human Neural Progenitor Cells Exposed to Lead (Pb) Reveals Transcriptome Dynamics, Splicing Alterations and Disease Risk Associations.Toxicol Sci. 2017 Sep 1;159(1):251-265. doi: 10.1093/toxsci/kfx129. Toxicol Sci. 2017. PMID: 28903495 Free PMC article.
-
Unraveling the Pathways to Neuronal Homeostasis and Disease: Mechanistic Insights into the Role of RNA-Binding Proteins and Associated Factors.Int J Mol Sci. 2018 Aug 3;19(8):2280. doi: 10.3390/ijms19082280. Int J Mol Sci. 2018. PMID: 30081499 Free PMC article. Review.
References
-
- Saito-Hisaminato A, Katagiri T, Kakiuchi S, Nakamura T, Tsunoda T, Nakamura Y. Genome-wide profiling of gene expression in 29 normal human tissues with a cDNA microarray. DNA Res. 2002;9:35–45. - PubMed
-
- Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006;126:37–47. - PubMed
-
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003;72:291–336. - PubMed
