

Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells
- PMID: 20110409
- PMCID: PMC2832136
- DOI: 10.2353/ajpath.2010.090832
Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells
Abstract
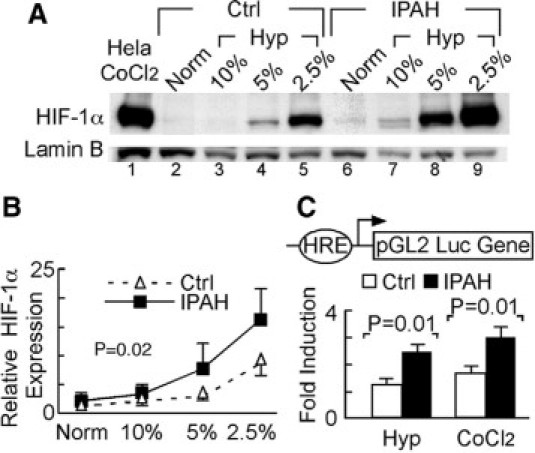
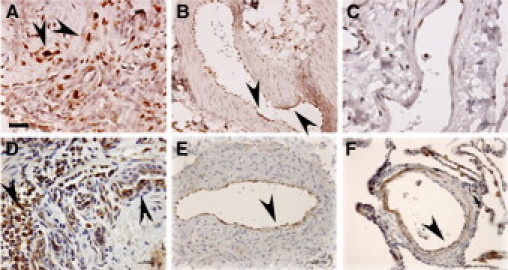
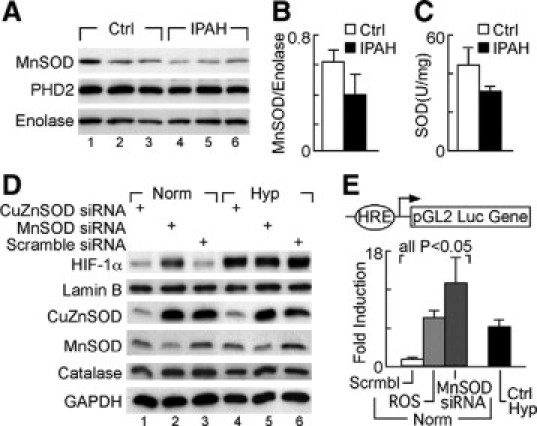
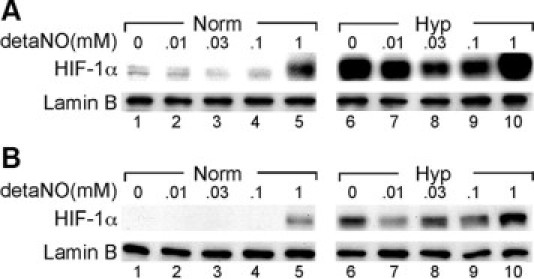
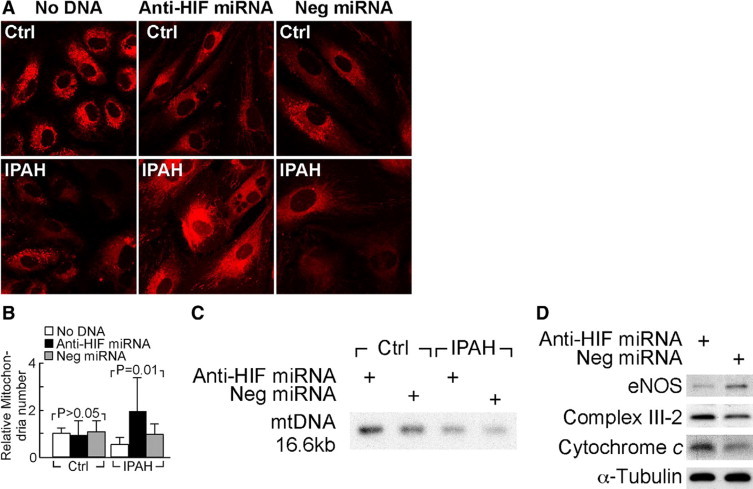
Severe pulmonary hypertension is irreversible and often fatal. Abnormal proliferation and resistance to apoptosis of endothelial cells (ECs) and hypertrophy of smooth muscle cells in this disease are linked to decreased mitochondria and preferential energy generation by glycolysis. We hypothesized this metabolic shift of pulmonary hypertensive ECs is due to greater hypoxia inducible-factor1alpha (HIF-1alpha) expression caused by low levels of nitric oxide combined with low superoxide dismutase activity. We show that cultured ECs from patients with idiopathic pulmonary arterial hypertension (IPAH-ECs) have greater HIF-1alpha expression and transcriptional activity than controls under normoxia or hypoxia, and pulmonary arteries from affected patients have increased expression of HIF-1alpha and its target carbonic anhydrase IX. Decreased expression of manganese superoxide dismutase (MnSOD) in IPAH-ECs paralleled increased HIF-1alpha levels and small interfering (SI) RNA knockdown of MnSOD, but not of the copper-zinc SOD, increased HIF-1 protein expression and hypoxia response element (HRE)-driven luciferase activity in normoxic ECs. MnSOD siRNA also reduced nitric oxide production in supernatants of IPAH-ECs. Conversely, low levels of a nitric oxide donor reduced HIF-1alpha expression in normoxic IPAH-ECs. Finally, mitochondria numbers increased in IPAH-ECs with knockdown of HIF-1alpha. These findings indicate that alterations of nitric oxide and MnSOD contribute to pathological HIF-1alpha expression and account for lower numbers of mitochondria in IPAH-ECs.
Figures
References
-
- Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67:737–744. - PMC - PubMed
-
- The International PPH Consortium, Lane KB, Machado RD, Pauciulo. M.W., Thompson JR, Philips JA, III, Loyd JE, Nichols WC, Trembath RC. Heterozygous germline mutations in BMPR2 encoding a TGF-B receptor cause familiar pulmonary hypertension. Nat Genet. 2000;26:81–84. - PubMed
-
- Voelkel NF, Cool CD, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1999;114:225S–230S. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
