

Review
doi: 10.1002/wnan.73.
New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders
Affiliations
- PMID: 20112244
- PMCID: PMC4002210
- DOI: 10.1002/wnan.73
Item in Clipboard
Review
New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders
Wiley Interdiscip Rev Nanomed Nanobiotechnol.
2010 Mar-Apr.
Abstract
This review discusses the multiple bio- and nanotechnological strategies developed in the last few decades for treatment of a group of fatal genetic diseases termed lysosomal storage disorders. Some basic foundation on the biomedical causes and social and clinical relevance of these diseases is provided. Several treatment modalities, from those currently available to novel therapeutic approaches under development, are also discussed; these include gene and cell therapies, substrate reduction therapy, chemical chaperones, enzyme replacement therapy, multifunctional chimeras, targeting strategies, and drug carrier approaches.
Figures
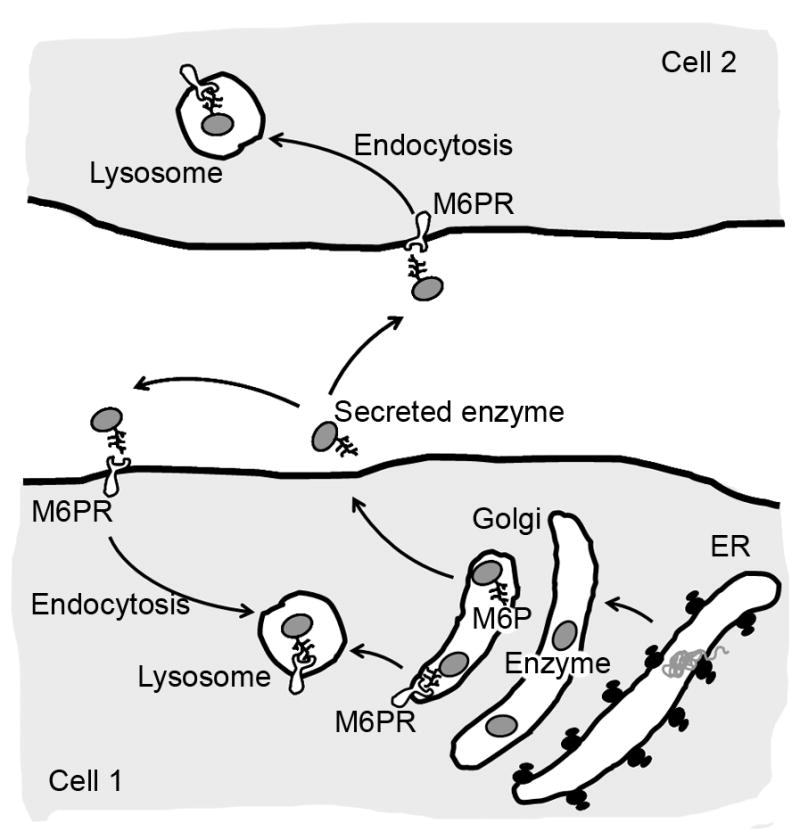
After their biosynthesis in the endoplasmic reticulum (ER), lysosomal enzymes are transported to the Golgi apparatus where they are modified by addition of sugar residues and, in some cases, phosphorylation. Certain enzyme residues, such as mannose 6 phosphate (M6P) are then recognized by M6P receptors (M6PR), which mediate intracellular trafficking to lysosomes. Lysosomal enzymes are also “secreted” to the extracellular milieu, where they can bind to M6PR on the cell surface of the same cell or a different cell. This mediates clathrin-mediated uptake and endocytic transport to lysosomes. This pathway constitutes the foundation for delivery of recombinant enzymes in replacement therapy protocols.
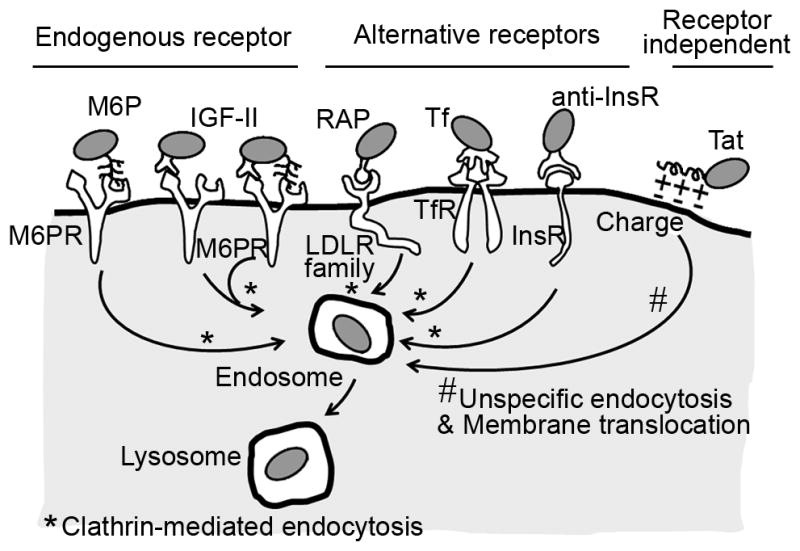
To improve delivery of replacement therapies for treatment of lysosomal storage disorders, the recombinant enzymes can be produced as multifunctional fusion proteins or chimeras comprising the enzymatic module and an affinity peptide, which provides affinity to the cell surface. These peptides can be designed to target receptors of endogenous lysosomal enzymes, such as the mannose 6 phosphate receptor (M6PR), which can be targeted by M6P and insulin-like growth factor II (IGF II). Chimeric enzymes can also be targeted to alternative receptors of endocytosis, such as low density lipoprotein receptor (LDLR) family family, which is targeted by receptor associated protein (RAP), transferrin receptor (TfR), targeted by transferrin (Tf), or insulin receptor (InsR), targeted by a peptide derived from an antibody to InsR (anti-InsR). These fusion enzymes can also be addressed to negatively charged domains of the plasma membrane, which can be targeted by Tat peptides. Chimeric enzymes targeted by these peptides can enter cells and traffic to lysosomes via clathrin-mediated endocytosis (those targeted to the receptors described above) or unspecific endocytosis (those targeted by Tat).
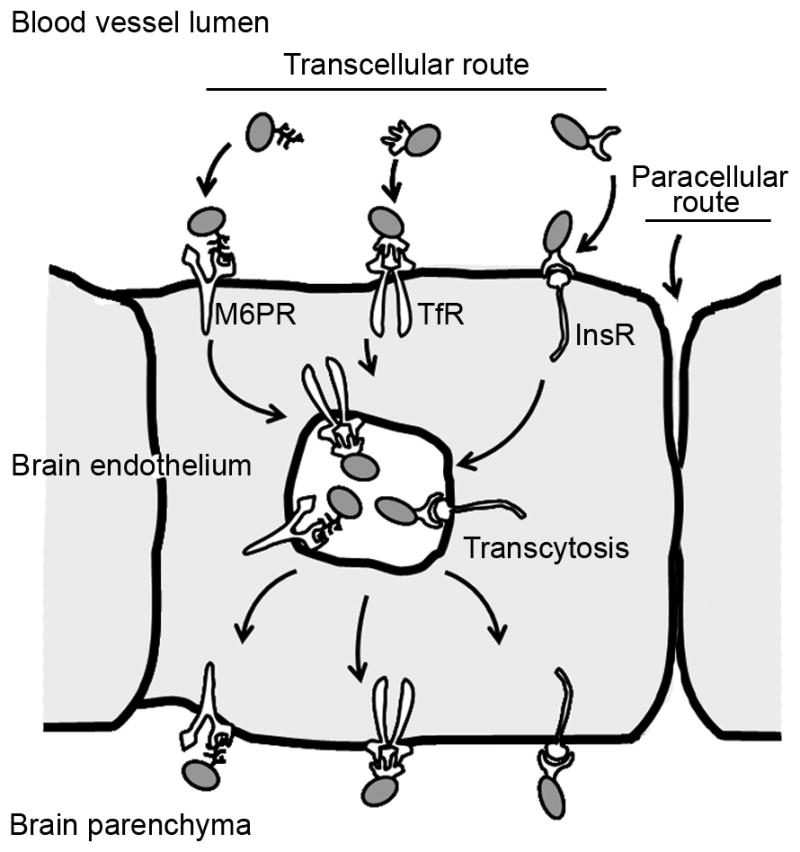
To improve delivery of replacement therapies to the brain in the case of lysosomal storage disorders affecting the central nervous system, recombinant enzymes can be designed to target receptors involved in transcellular transport of substances across the endothelium in the blood-brain barrier. This transport mediated by vesicular transcytosis is relatively safe, as it does not cause opening of the junctions between adjacent endothelial cells maintaining the permeability barrier (the paracellular route). M6PR = mannose 6 phosphate receptor; TfR = transferrin receptor; InsR = insulin receptor.
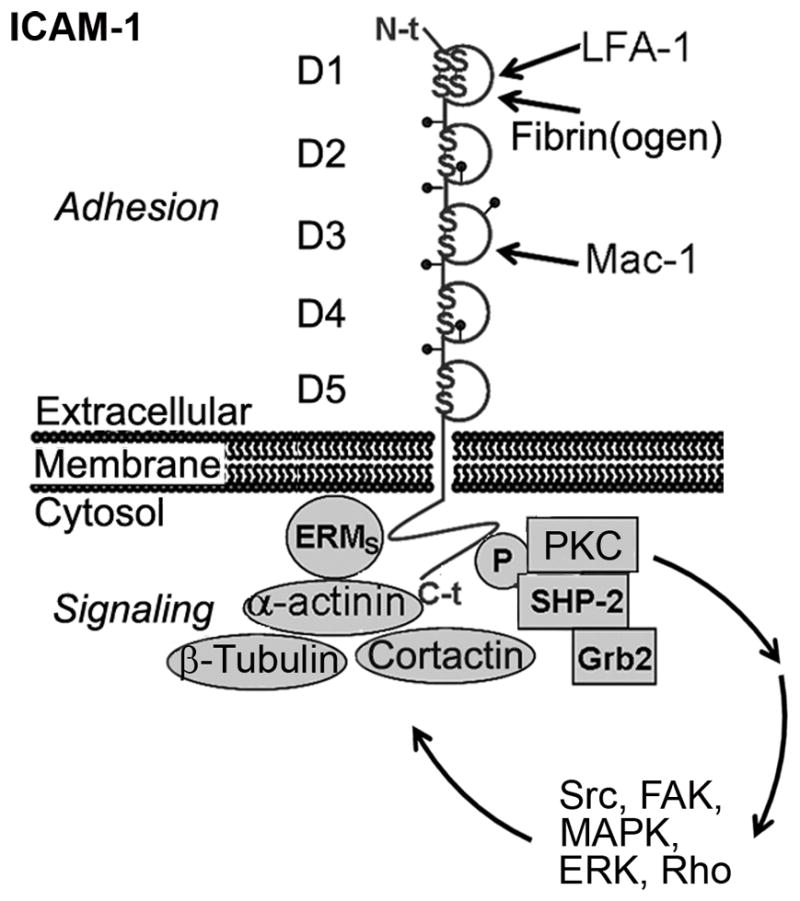
ICAM-1 is characterized by a long glycosylated extracellular region with five immunoglobulin-like domains (D1 to D5), a transmembrane region, and a short cytosolic tail. The extracellular domains mediate interaction with leukocyte β2 integrins (LFA and Mac-1) and fibrin(ogen). The cytosolic domain mediates signal transduction upon engagement of the extracellular domains. ICAM-1-mediated signaling includes tyrosine phosphorylation of SHP-2 helped by Grb2, PKC activation, phosphorylation of Src kinases, signaling through MAP kinases, Rho-dependent remodeling of the cytoskeleton, activation of cortactin and focal adhesion kinase (FAK), and interaction with α-actinin, ezrin and moesin, and the microtubule-related protein β-tubulin [95].
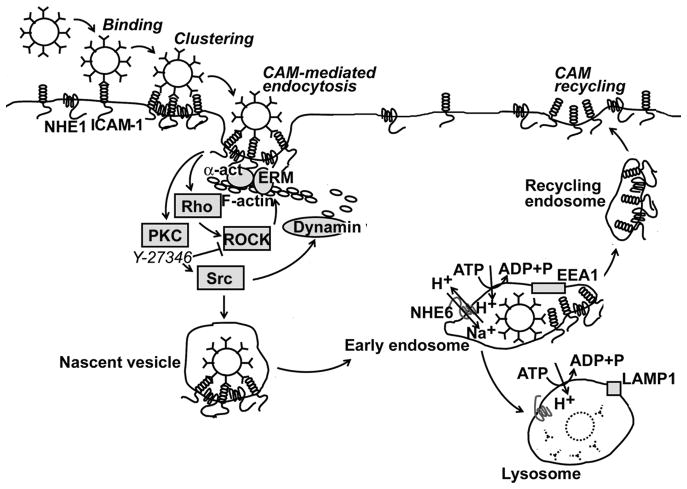
CAM-endocytosis is induced upon ICAM-1 engagement by multivalent ligands, such as 100 nm polystyrene or PLGA nanocarriers coated by anti-ICAM. This pathway involves PKC, Src kinases, and Rho-dependent kinase (ROCK) and formation of actin stress fibers. ICAM-1 interacts with the amiloride-sensitive, sodium/proton exchanger NHE1, which contributes to cell surface deformability and is an adaptor of the cytoskeleton. Dynamin is involved in budding of the nascent vesicles. These events contribute to the progression of membrane invaginations and intracellular vesicular transport. After internalization, ICAM-1/anti-ICAM carrier complexes traffic to endosomes that are positive for EEA1 and PKC-regulated NHE6. ICAM-1 recycles then to the plasmalemma, and anti-ICAM nanocarriers traffic to LAMP1-positive lysosomes.
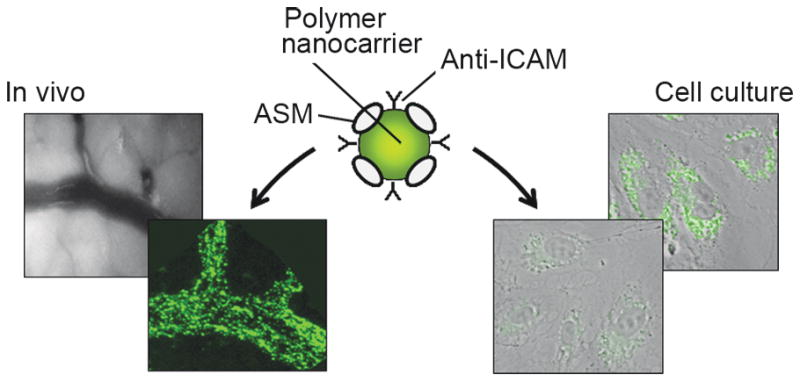
Model polymer nanocarriers (FITC-labeled polystyrene, 100 nm diameter) were coated by surface absorption with recombinant acid sphingomyelinase (ASM, Schuchman – Mount Sinai School of Medicine) and monoclonal anti-ICAM or control IgG. Upon injection in vivo in mice, only ICAM-1-targeted carriers (lower panel, left), but not control IgG carriers (upper panel, left), showed efficient binding to the vasculature irrigating all organs (mesentery is shown) [117]. At the cellular level, incubation of skin fibroblasts from type B Niemann-Pick disease patients with non-fluorescent carriers for 3 h (lower panel, right) significantly attenuated lysosomal sphingomyelin (labeled by a green-fluorescent BODIPY derivative, upper panel, right) [105].
Similar articles
-
Treatment strategies for lysosomal storage disorders.Dev Med Child Neurol. 2018 Jan;60(1):13-18. doi: 10.1111/dmcn.13600. Epub 2017 Nov 1. Dev Med Child Neurol. 2018. PMID: 29090451 Review.
-
Lysosomal storage diseases: from pathophysiology to therapy.Annu Rev Med. 2015;66:471-86. doi: 10.1146/annurev-med-122313-085916. Annu Rev Med. 2015. PMID: 25587658 Review.
-
Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923-2016).J Inherit Metab Dis. 2017 May;40(3):343-356. doi: 10.1007/s10545-017-0032-8. Epub 2017 Mar 17. J Inherit Metab Dis. 2017. PMID: 28314976 Review.
-
New strategies for the treatment of lysosomal storage diseases (review).Int J Mol Med. 2013 Jan;31(1):11-20. doi: 10.3892/ijmm.2012.1187. Epub 2012 Nov 19. Int J Mol Med. 2013. PMID: 23165354 Review.
-
Therapeutic Approaches in Lysosomal Storage Diseases.Biomolecules. 2021 Nov 26;11(12):1775. doi: 10.3390/biom11121775. Biomolecules. 2021. PMID: 34944420 Free PMC article. Review.
Cited by
-
Flow shear stress differentially regulates endothelial uptake of nanocarriers targeted to distinct epitopes of PECAM-1.J Control Release. 2015 Jul 28;210:39-47. doi: 10.1016/j.jconrel.2015.05.006. Epub 2015 May 9. J Control Release. 2015. PMID: 25966362 Free PMC article.
-
Biocompatible Polymer Nanoparticles for Drug Delivery Applications in Cancer and Neurodegenerative Disorder Therapies.J Funct Biomater. 2019 Jan 8;10(1):4. doi: 10.3390/jfb10010004. J Funct Biomater. 2019. PMID: 30626094 Free PMC article. Review.
-
Targeted enzyme delivery systems in lysosomal disorders: an innovative form of therapy for mucopolysaccharidosis.Cell Mol Life Sci. 2019 Sep;76(17):3363-3381. doi: 10.1007/s00018-019-03135-z. Epub 2019 May 17. Cell Mol Life Sci. 2019. PMID: 31101939 Free PMC article. Review.
-
Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases.Autophagy. 2016;12(3):472-83. doi: 10.1080/15548627.2015.1136769. Autophagy. 2016. PMID: 26761717 Free PMC article.
-
Application of advances in endocytosis and membrane trafficking to drug delivery.Adv Drug Deliv Rev. 2020;157:118-141. doi: 10.1016/j.addr.2020.07.026. Epub 2020 Aug 3. Adv Drug Deliv Rev. 2020. PMID: 32758615 Free PMC article. Review.
References
-
- Sabatini DD, Adesnik MB. The biogenesis of membranes and organelles. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, editors. Lysosomal disorders the metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 433–520.
-
- Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001.
-
- Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–565. - PubMed
-
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. Jama. 1999;281:249–254. - PubMed
-
- Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: Acid sphingomyelinase deficiencies. In: Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B, editors. Lysosomal disorders the metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3589–3610.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
