

Value of using multiple proteases for large-scale mass spectrometry-based proteomics
- PMID: 20113005
- PMCID: PMC2833215
- DOI: 10.1021/pr900863u
Value of using multiple proteases for large-scale mass spectrometry-based proteomics
Abstract
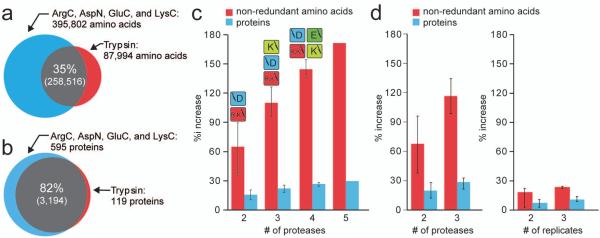
Large-scale protein sequencing methods rely on enzymatic digestion of complex protein mixtures to generate a collection of peptides for mass spectrometric analysis. Here we examine the use of multiple proteases (trypsin, LysC, ArgC, AspN, and GluC) to improve both protein identification and characterization in the model organism Saccharomyces cerevisiae. Using a data-dependent, decision tree-based algorithm to tailor MS(2) fragmentation method to peptide precursor, we identified 92 095 unique peptides (609 665 total) mapping to 3908 proteins at a 1% false discovery rate (FDR). These results were a significant improvement upon data from a single protease digest (trypsin) - 27 822 unique peptides corresponding to 3313 proteins. The additional 595 protein identifications were mainly from those at low abundances (i.e., < 1000 copies/cell); sequence coverage for these proteins was likewise improved nearly 3-fold. We demonstrate that large portions of the proteome are simply inaccessible following digestion with a single protease and that multiple proteases, rather than technical replicates, provide a direct route to increase both protein identifications and proteome sequence coverage.
Figures




References
-
- Washburn MP, Wolters D, Yates JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotech. 2001;19(3):242–247. - PubMed
-
- de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455(7217):1251–1254. - PubMed
-
- Peng JM, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: The yeast proteome. Journal of Proteome Research. 2003;2(1):43–50. - PubMed
-
- Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. Journal of the American Chemical Society. 1996;118(35):8365–8374.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
