

Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia
- PMID: 20116044
- PMCID: PMC2820177
- DOI: 10.1016/j.ajhg.2009.12.015
Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond-Blackfan anemia
Erratum in
- Am J Hum Genet. 2010 Apr 9;86(4):655
Abstract
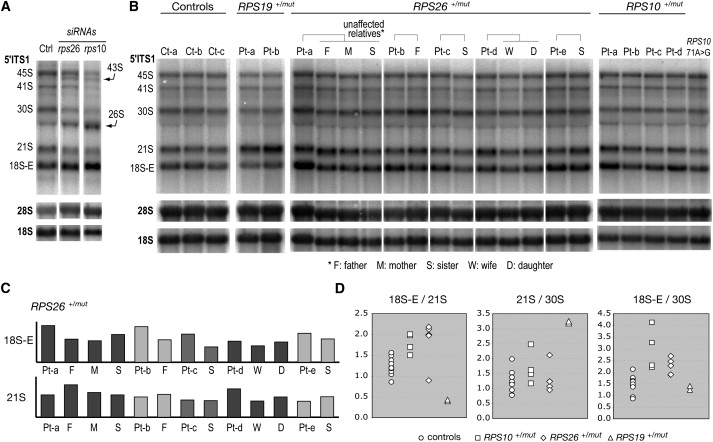
Diamond-Blackfan anemia (DBA), an inherited bone marrow failure syndrome characterized by anemia that usually presents before the first birthday or in early childhood, is associated with birth defects and an increased risk of cancer. Although anemia is the most prominent feature of DBA, the disease is also characterized by growth retardation and congenital malformations, in particular craniofacial, upper limb, heart, and urinary system defects that are present in approximately 30%-50% of patients. DBA has been associated with mutations in seven ribosomal protein (RP) genes, RPS19, RPS24, RPS17, RPL35A, RPL5, RPL11, and RPS7, in about 43% of patients. To continue our large-scale screen of RP genes in a DBA population, we sequenced 35 ribosomal protein genes, RPL15, RPL24, RPL29, RPL32, RPL34, RPL9, RPL37, RPS14, RPS23, RPL10A, RPS10, RPS12, RPS18, RPL30, RPS20, RPL12, RPL7A, RPS6, RPL27A, RPLP2, RPS25, RPS3, RPL41, RPL6, RPLP0, RPS26, RPL21, RPL36AL, RPS29, RPL4, RPLP1, RPL13, RPS15A, RPS2, and RPL38, in our DBA patient cohort of 117 probands. We identified three distinct mutations of RPS10 in five probands and nine distinct mutations of RPS26 in 12 probands. Pre-rRNA analysis in lymphoblastoid cells from patients bearing mutations in RPS10 and RPS26 showed elevated levels of 18S-E pre-rRNA. This accumulation is consistent with the phenotype observed in HeLa cells after knockdown of RPS10 or RPS26 expression with siRNAs, which indicates that mutations in the RPS10 and RPS26 genes in DBA patients affect the function of the proteins in rRNA processing.
Copyright (c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Alter B.P., Young N.S. The bone marrow failure syndromes. In: Nathan D.G., Orkin H.S., editors. Hematology of Infancy and Childhood. Volume 1. Saunders; Philadelphia: 1998. pp. 237–335.
-
- Willig T.N., Niemeyer C.M., Leblanc T., Tiemann C., Robert A., Budde J., Lambiliotte A., Kohne E., Souillet G., Eber S. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d'Hématologie et d'Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr. Res. 1999;46:553–561. - PubMed
-
- Orfali K.A., Ohene-Abuakwa Y., Ball S.E. Diamond Blackfan anaemia in the UK: Clinical and genetic heterogeneity. Br. J. Haematol. 2004;125:243–252. - PubMed
-
- Lipton J.M., Atsidaftos E., Zyskind I., Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: An update from the Diamond Blackfan Anemia Registry. Pediatr. Blood Cancer. 2006;46:558–564. - PubMed
-
- Janov A.J., Leong T., Nathan D.G., Guinan E.C. Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine (Baltimore) 1996;75:77–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
