

Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry
- PMID: 20116280
- PMCID: PMC2901854
- DOI: 10.1016/j.jasms.2009.12.016
Automated data reduction for hydrogen/deuterium exchange experiments, enabled by high-resolution Fourier transform ion cyclotron resonance mass spectrometry
Abstract
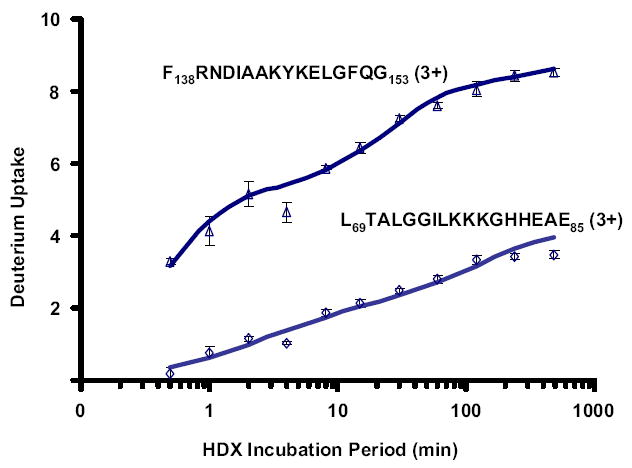
Mass analysis of proteolytic fragment peptides following hydrogen/deuterium exchange offers a general measure of solvent accessibility/hydrogen bonding (and thus conformation) of solution-phase proteins and their complexes. The primary problem in such mass analyses is reliable and rapid assignment of mass spectral peaks to the correct charge state and degree of deuteration of each fragment peptide, in the presence of substantial overlap between isotopic distributions of target peptides, autolysis products, and other interferant species. Here, we show that at sufficiently high mass resolving power (m/Delta m(50%) > or = 100,000), it becomes possible to resolve enough of those overlaps so that automated data reduction becomes possible, based on the actual elemental composition of each peptide without the need to deconvolve isotopic distributions. We demonstrate automated, rapid, reliable assignment of peptide masses from H/D exchange experiments, based on electrospray ionization FT-ICR mass spectra from H/D exchange of solution-phase myoglobin. Combined with previously demonstrated automated data acquisition for such experiments, the present data reduction algorithm enhances automation (and thus expands generality and applicability) for high-resolution mass spectrometry-based analysis of H/D exchange of solution-phase proteins.
2010 American Society for Mass Spectrometry. Published by Elsevier Inc. All rights reserved.
Figures
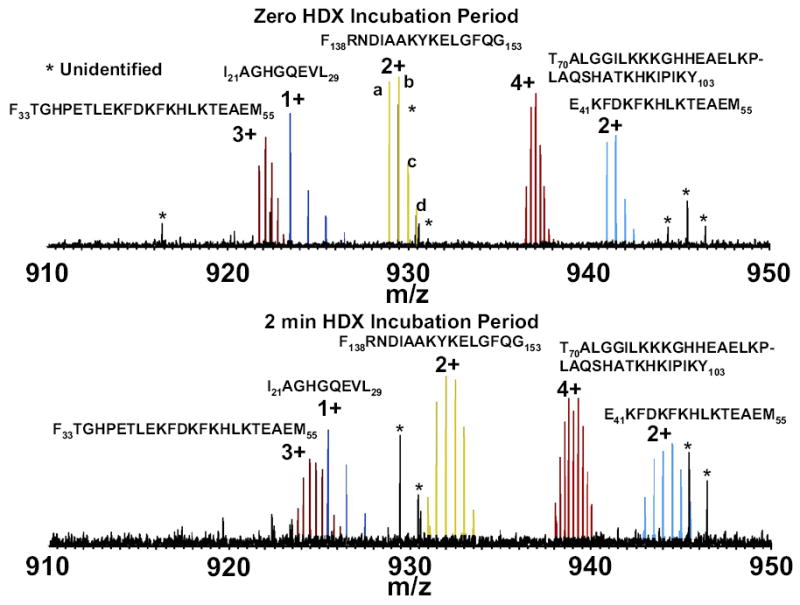
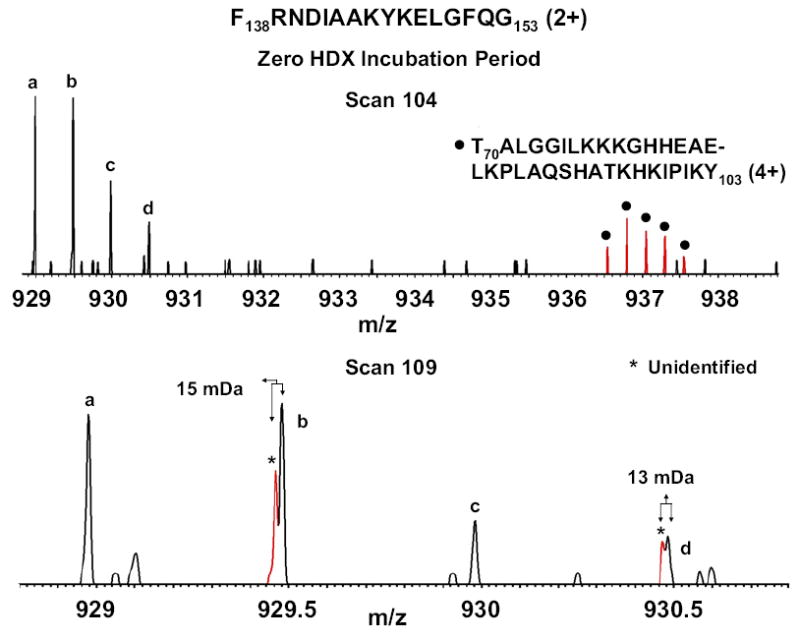
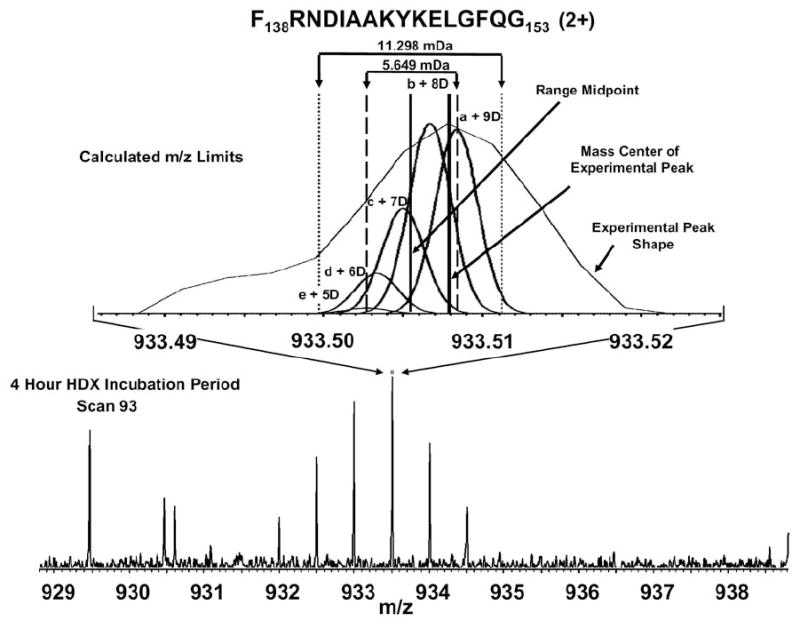
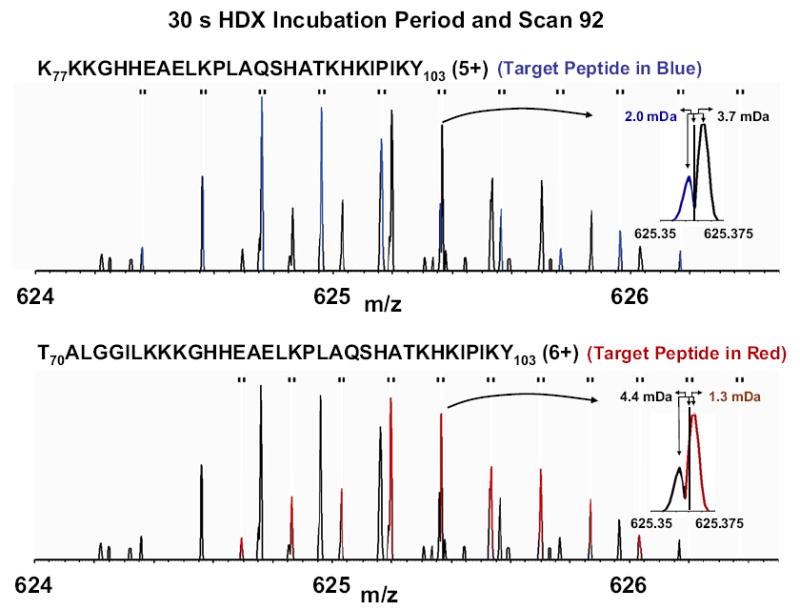
References
-
- Truhlar SME, Droy CH, Torpey JW, Koeppe JR, Komives EA. Solvent Accessibility of Protein Surfaces by Amide H/2H Exchange MALDI-TOF Mass Spectrometry. J Am Soc Mass Spectrom. 2006;17:1490–1497. - PubMed
-
- Weis DD, Wales TE, Engen JR, Hotchko M, Eyck LFT. Identification and Characterization of EX1 Kinetics in H/D Exchange Mass Spectrometry by Peak Width Analysis. J Am Soc Mass Spectrom. 2006;17:1498–1509. - PubMed
-
- Wang F, Li W, Emmett MR, Hendrickson CL, Marshall AG, Zhang YL, Wu L, Zhang ZY. Conformational and Dynamic Changes of Yersinia Protein Tyrosine Phosphatase Induced by Ligand Binding and Active Site Mutation and Revealed by H/D Exchange and Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Biochemistry. 1998;37:15289–15299. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
