

Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy
- PMID: 20117437
- PMCID: PMC3000630
- DOI: 10.1016/j.jacc.2009.11.017
Familial dilated cardiomyopathy caused by an alpha-tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy
Abstract
Objectives: We sought to further define the role of sarcomere mutations in dilated cardiomyopathy (DCM) and associated clinical phenotypes.
Background: Mutations in several contractile proteins contribute to DCM, but definitive evidence for the roles of most sarcomere genes remains limited by the lack of robust genetic support.
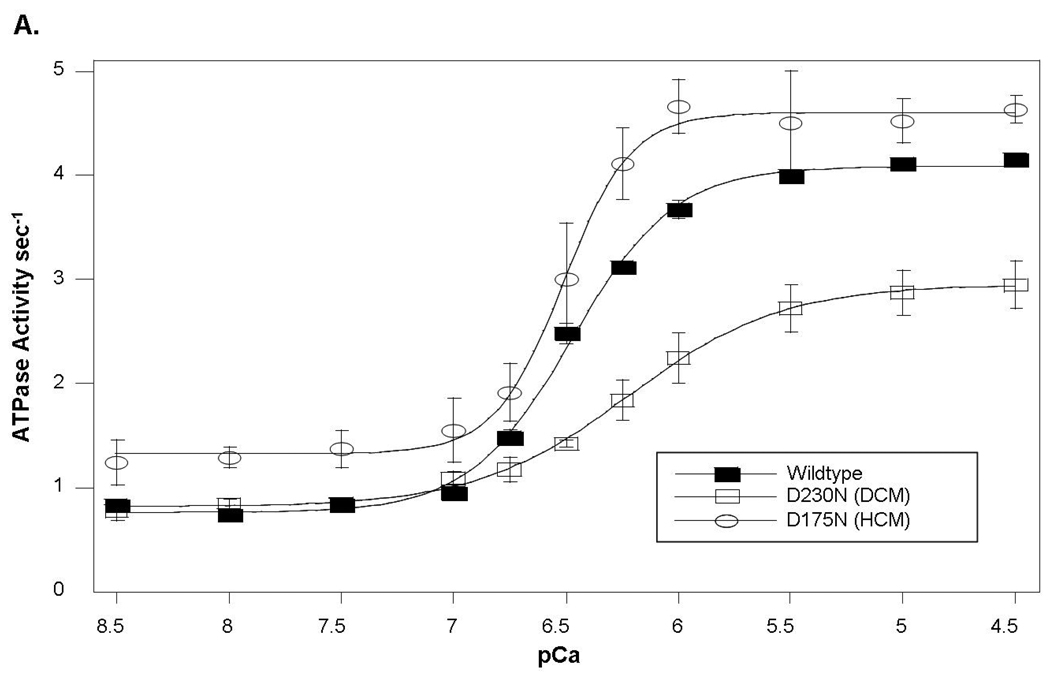
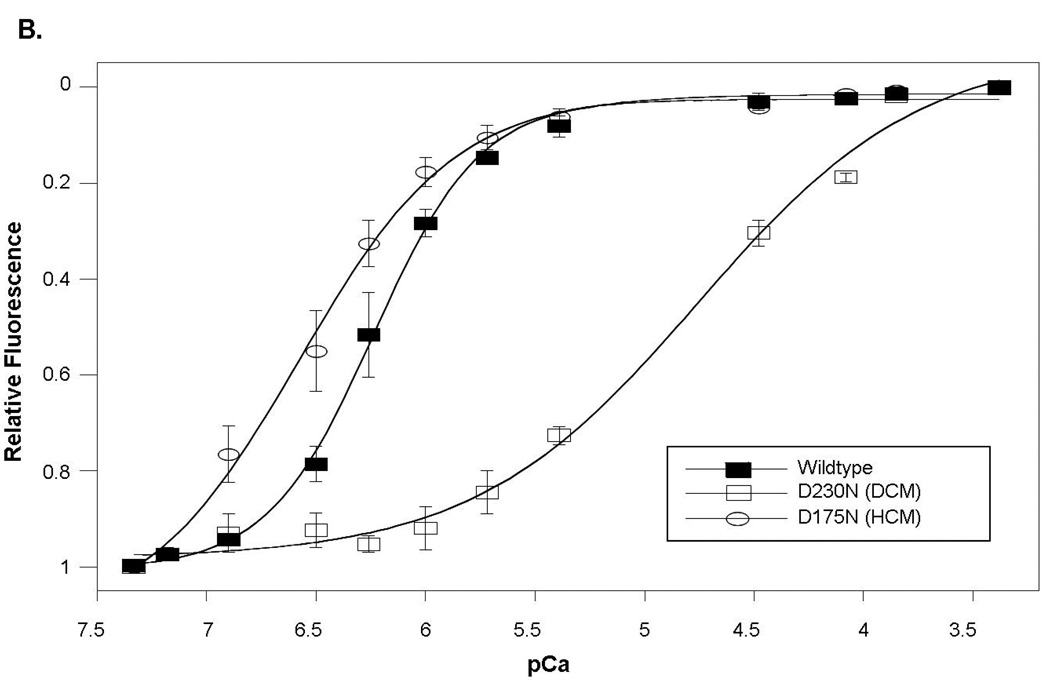
Methods: Direct sequencing of 6 sarcomere genes was performed on 334 probands with DCM. A novel D230N missense mutation in the gene encoding alpha-tropomyosin (TPM1) was identified. Functional assessment was performed by the use of an in vitro reconstituted sarcomere complex to evaluate ATPase regulation and Ca(2+) affinity as correlates of contractility.
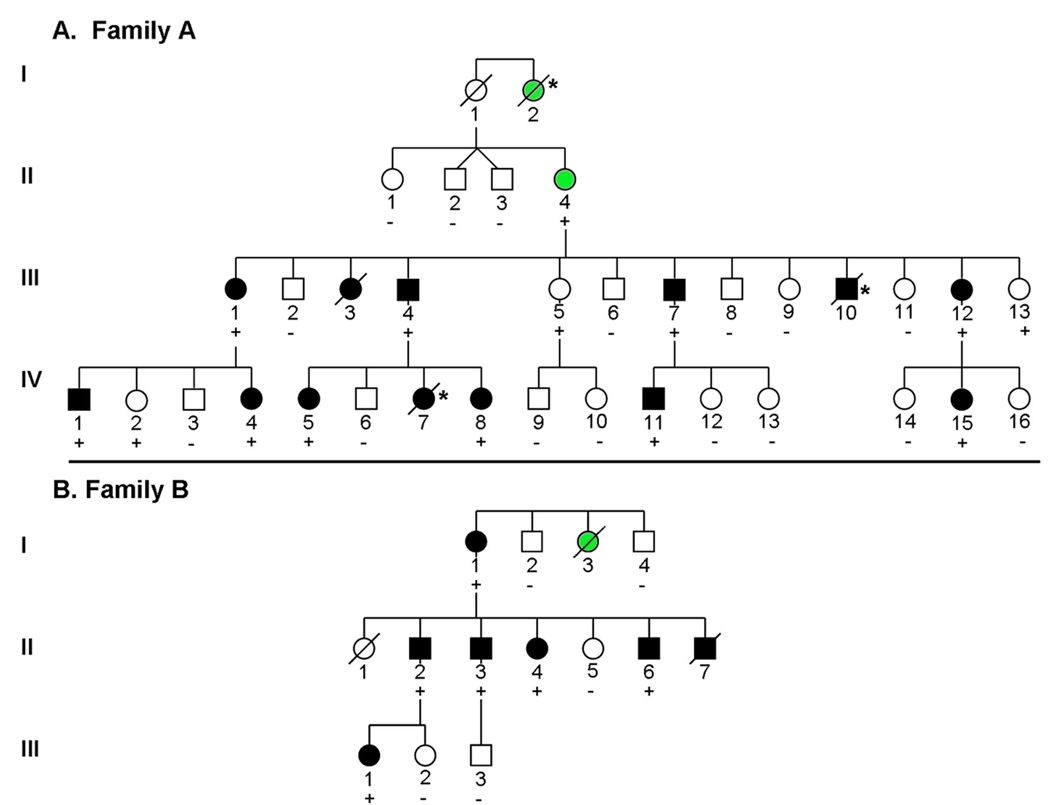
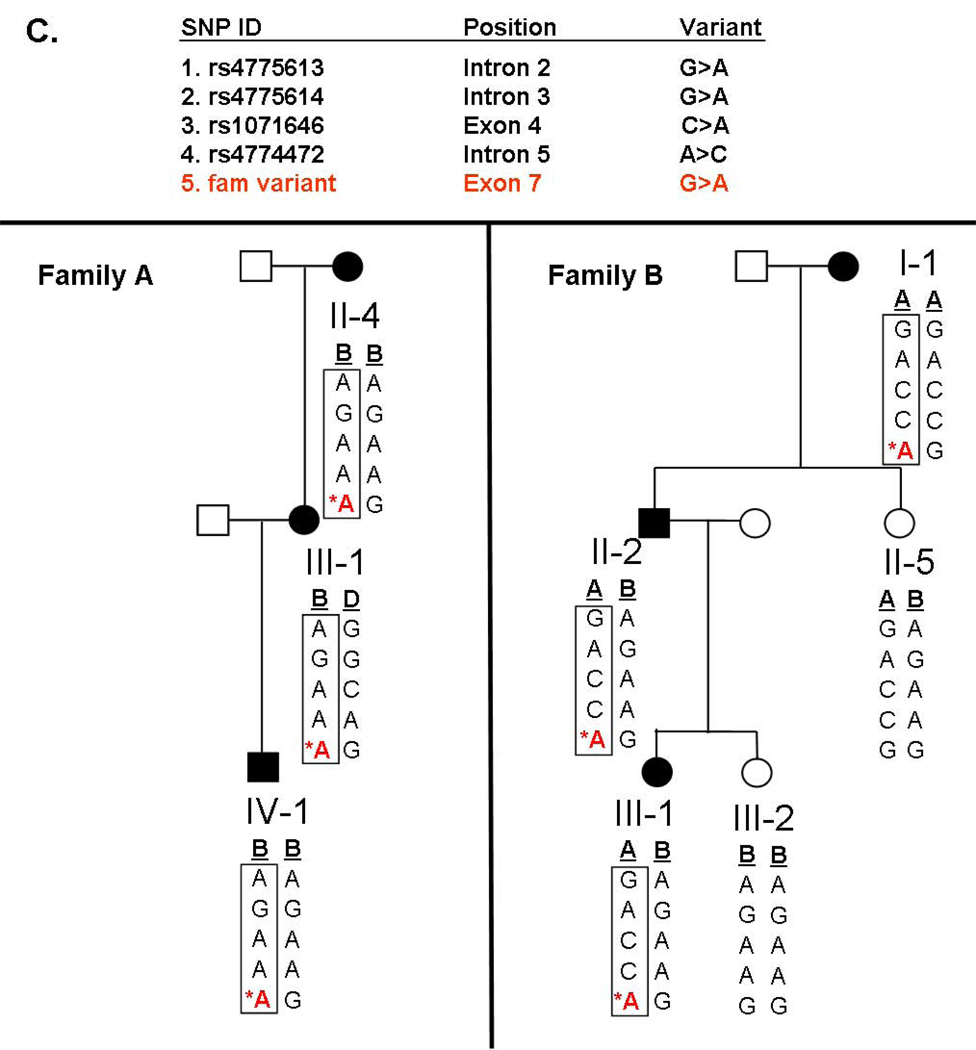
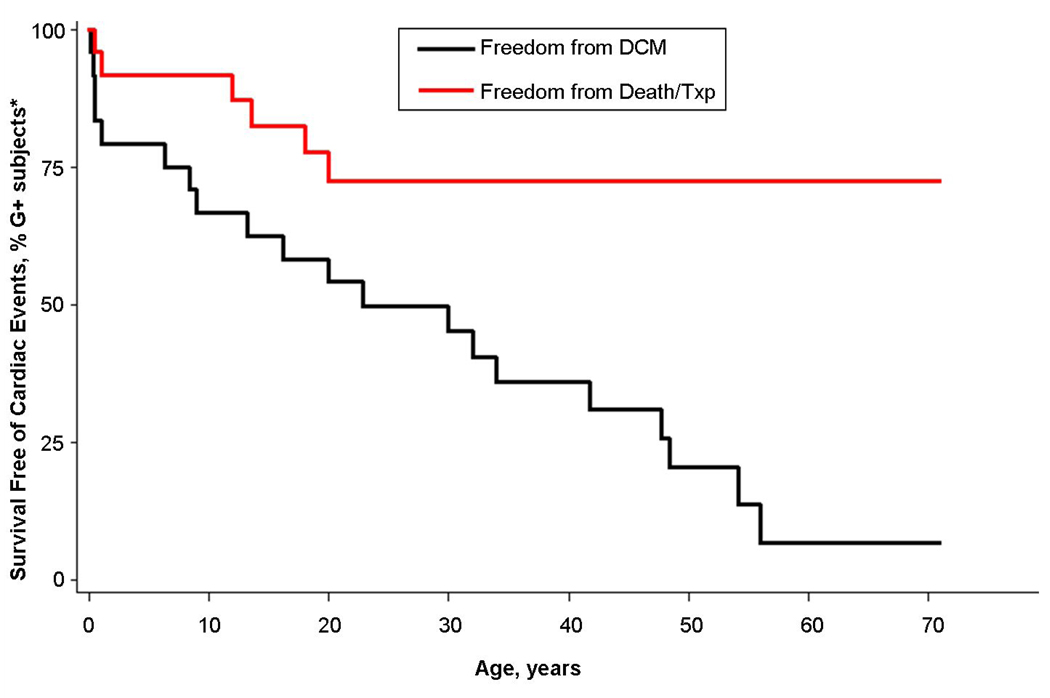
Results: TPM1 D230N segregated with DCM in 2 large unrelated families. This mutation altered an evolutionarily conserved residue and was absent in >1,000 control chromosomes. In vitro studies demonstrated major inhibitory effects on sarcomere function with reduced Ca(2+) sensitivity, maximum activation, and Ca(2+) affinity compared with wild-type TPM1. Clinical manifestations ranged from decompensated heart failure or sudden death in those presenting early in life to asymptomatic left ventricular dysfunction in those diagnosed during adulthood. Notably, several affected infants had remarkable improvement.
Conclusions: Genetic segregation in 2 unrelated families and functional analyses conclusively establish a pathogenic role for TPM1 mutations in DCM. In vitro results demonstrate contrasting effects of DCM and hypertrophic cardiomyopathy mutations in TPM1, suggesting that specific functional consequences shape cardiac remodeling. Along with previous reports, our data support a distinctive, age-dependent phenotype with sarcomere-associated DCM where presentation early in life is associated with severe, sometimes lethal, disease. These observations have implications for the management of familial DCM.
Copyright (c) 2010 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors have no conflicts of interest to report.
Figures
Comment in
-
Tropomyosin and dilated cardiomyopathy: revenge of the actinomyosin "gatekeeper".J Am Coll Cardiol. 2010 Jan 26;55(4):330-2. doi: 10.1016/j.jacc.2009.11.018. J Am Coll Cardiol. 2010. PMID: 20117438 No abstract available.
References
-
- Boucek MM, Aurora P, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Tenth Official Pediatric Heart Transplantation Report--2007. The Journal of Heart and Lung Transplantation. 2007;26:796–807. - PubMed
-
- Taylor DO, Edwards LB, Boucek MM, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report--2007. J Heart Lung Transplant. 2007;26:769–781. - PubMed
-
- Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–981. - PubMed
-
- Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. - PubMed
-
- Ho CY, Seidman CE. A Contemporary Approach to Hypertrophic Cardiomyopathy. Circulation. 2006;113:e858–e862. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
