

Prediction of protein structural classes for low-homology sequences based on predicted secondary structure
- PMID: 20122246
- PMCID: PMC3009544
- DOI: 10.1186/1471-2105-11-S1-S9
Prediction of protein structural classes for low-homology sequences based on predicted secondary structure
Abstract
Background: Prediction of protein structural classes (alpha, beta, alpha + beta and alpha/beta) from amino acid sequences is of great importance, as it is beneficial to study protein function, regulation and interactions. Many methods have been developed for high-homology protein sequences, and the prediction accuracies can achieve up to 90%. However, for low-homology sequences whose average pairwise sequence identity lies between 20% and 40%, they perform relatively poorly, yielding the prediction accuracy often below 60%.
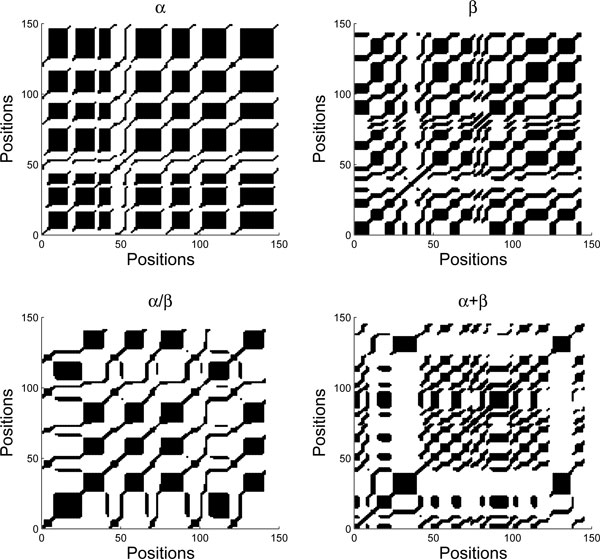
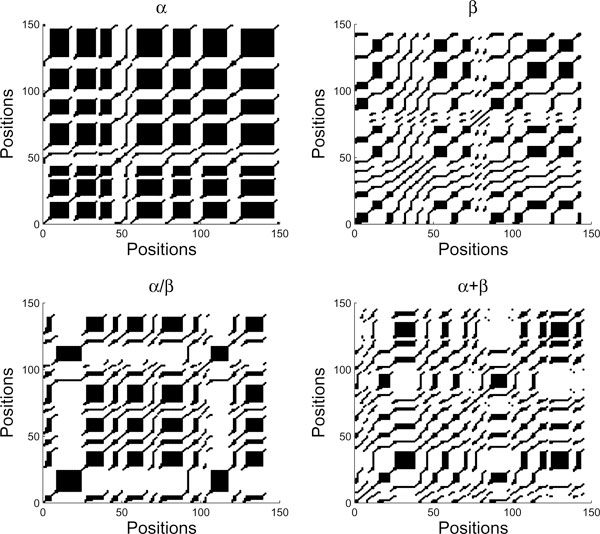
Results: We propose a new method to predict protein structural classes on the basis of features extracted from the predicted secondary structures of proteins rather than directly from their amino acid sequences. It first uses PSIPRED to predict the secondary structure for each protein sequence. Then, the chaos game representation is employed to represent the predicted secondary structure as two time series, from which we generate a comprehensive set of 24 features using recurrence quantification analysis, K-string based information entropy and segment-based analysis. The resulting feature vectors are finally fed into a simple yet powerful Fisher's discriminant algorithm for the prediction of protein structural classes. We tested the proposed method on three benchmark datasets in low homology and achieved the overall prediction accuracies of 82.9%, 83.1% and 81.3%, respectively. Comparisons with ten existing methods showed that our method consistently performs better for all the tested datasets and the overall accuracy improvements range from 2.3% to 27.5%. A web server that implements the proposed method is freely available at http://www1.spms.ntu.edu.sg/~chenxin/RKS_PPSC/.
Conclusion: The high prediction accuracy achieved by our proposed method is attributed to the design of a comprehensive feature set on the predicted secondary structure sequences, which is capable of characterizing the sequence order information, local interactions of the secondary structural elements, and spacial arrangements of alpha helices and beta strands. Thus, it is a valuable method to predict protein structural classes particularly for low-homology amino acid sequences.
Figures



Similar articles
-
Accurate prediction of protein structural classes by incorporating predicted secondary structure information into the general form of Chou's pseudo amino acid composition.J Theor Biol. 2014 Mar 7;344:12-8. doi: 10.1016/j.jtbi.2013.11.021. Epub 2013 Dec 6. J Theor Biol. 2014. PMID: 24316044
-
Prediction of protein structural classes by recurrence quantification analysis based on chaos game representation.J Theor Biol. 2009 Apr 21;257(4):618-26. doi: 10.1016/j.jtbi.2008.12.027. Epub 2009 Jan 8. J Theor Biol. 2009. PMID: 19183559
-
Prediction of protein structural class using novel evolutionary collocation-based sequence representation.J Comput Chem. 2008 Jul 30;29(10):1596-604. doi: 10.1002/jcc.20918. J Comput Chem. 2008. PMID: 18293306
-
Extending the Horizon of Homology Detection with Coevolution-based Structure Prediction.J Mol Biol. 2021 Oct 1;433(20):167106. doi: 10.1016/j.jmb.2021.167106. Epub 2021 Jun 15. J Mol Biol. 2021. PMID: 34139218 Free PMC article. Review.
-
Comparison of proteins based on segments structural similarity.Acta Biochim Pol. 2004;51(1):161-72. Acta Biochim Pol. 2004. PMID: 15094837 Review.
Cited by
-
Customised fragments libraries for protein structure prediction based on structural class annotations.BMC Bioinformatics. 2015 Apr 29;16(1):136. doi: 10.1186/s12859-015-0576-2. BMC Bioinformatics. 2015. PMID: 25925397 Free PMC article.
-
Using Recursive Feature Selection with Random Forest to Improve Protein Structural Class Prediction for Low-Similarity Sequences.Comput Math Methods Med. 2021 May 7;2021:5529389. doi: 10.1155/2021/5529389. eCollection 2021. Comput Math Methods Med. 2021. PMID: 34055035 Free PMC article.
-
The HER2 target for designing novel multi-peptide vaccine against breast cancer using immunoinformatics and molecular dynamic simulation.Biochem Biophys Rep. 2025 Jul 4;43:102135. doi: 10.1016/j.bbrep.2025.102135. eCollection 2025 Sep. Biochem Biophys Rep. 2025. PMID: 40688509 Free PMC article.
-
Accurate prediction of protein structural class.PLoS One. 2012;7(6):e37653. doi: 10.1371/journal.pone.0037653. Epub 2012 Jun 19. PLoS One. 2012. PMID: 22723837 Free PMC article.
-
Comparison study on statistical features of predicted secondary structures for protein structural class prediction: From content to position.BMC Bioinformatics. 2013 May 4;14:152. doi: 10.1186/1471-2105-14-152. BMC Bioinformatics. 2013. PMID: 23641706 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
