

Identification of recurring protein structure microenvironments and discovery of novel functional sites around CYS residues
- PMID: 20122268
- PMCID: PMC2833161
- DOI: 10.1186/1472-6807-10-4
Identification of recurring protein structure microenvironments and discovery of novel functional sites around CYS residues
Abstract
Background: The emergence of structural genomics presents significant challenges in the annotation of biologically uncharacterized proteins. Unfortunately, our ability to analyze these proteins is restricted by the limited catalog of known molecular functions and their associated 3D motifs.
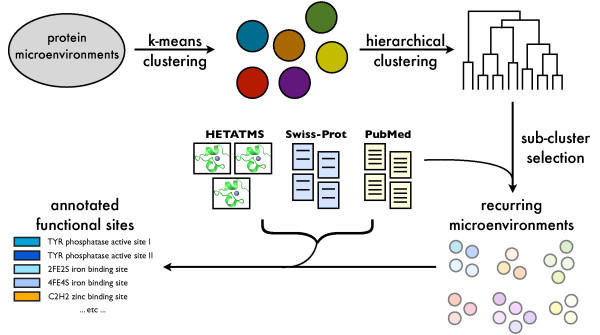
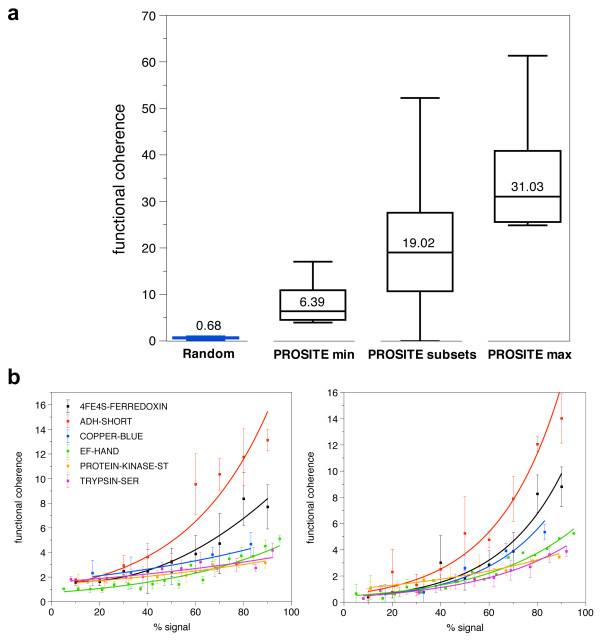
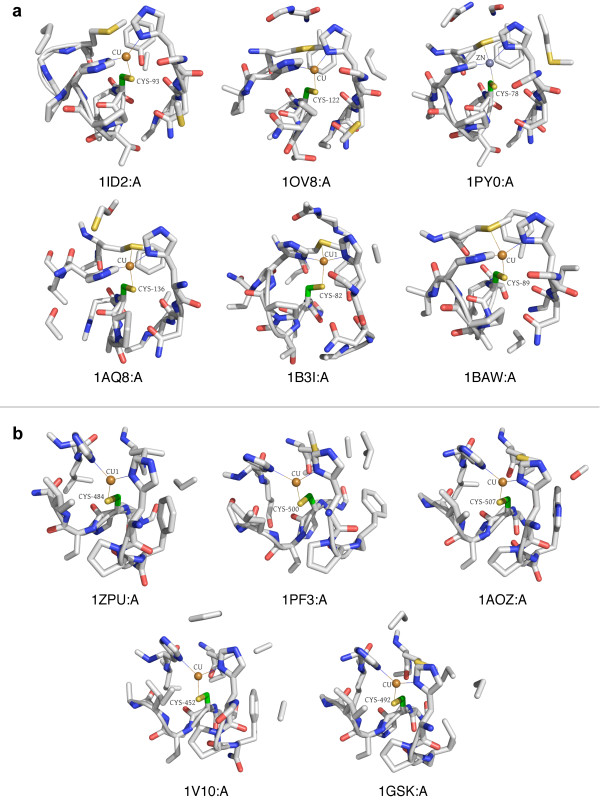
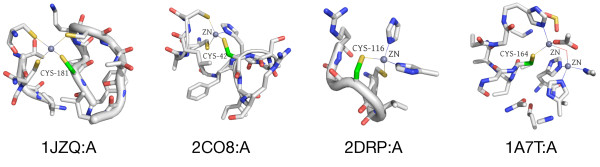
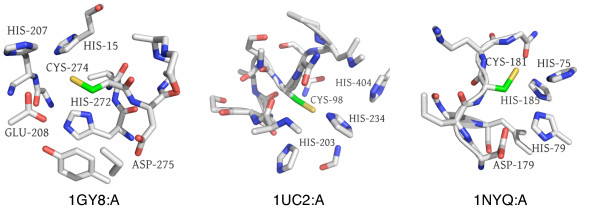
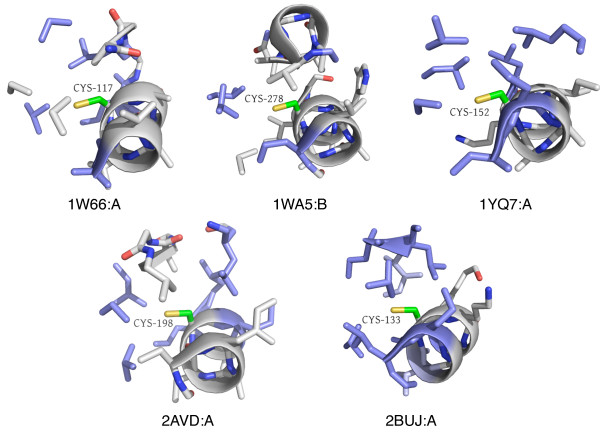
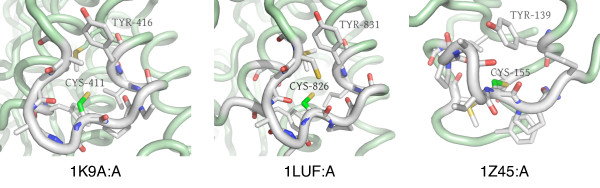
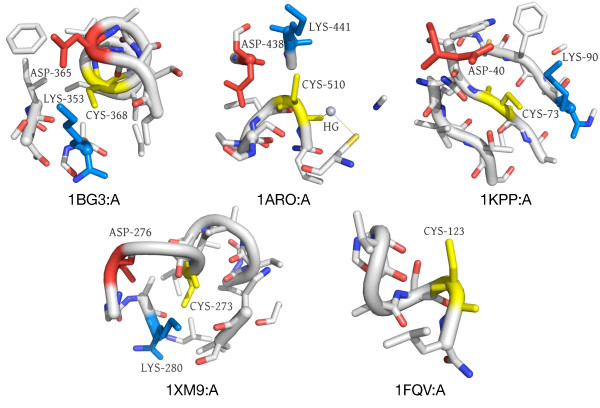
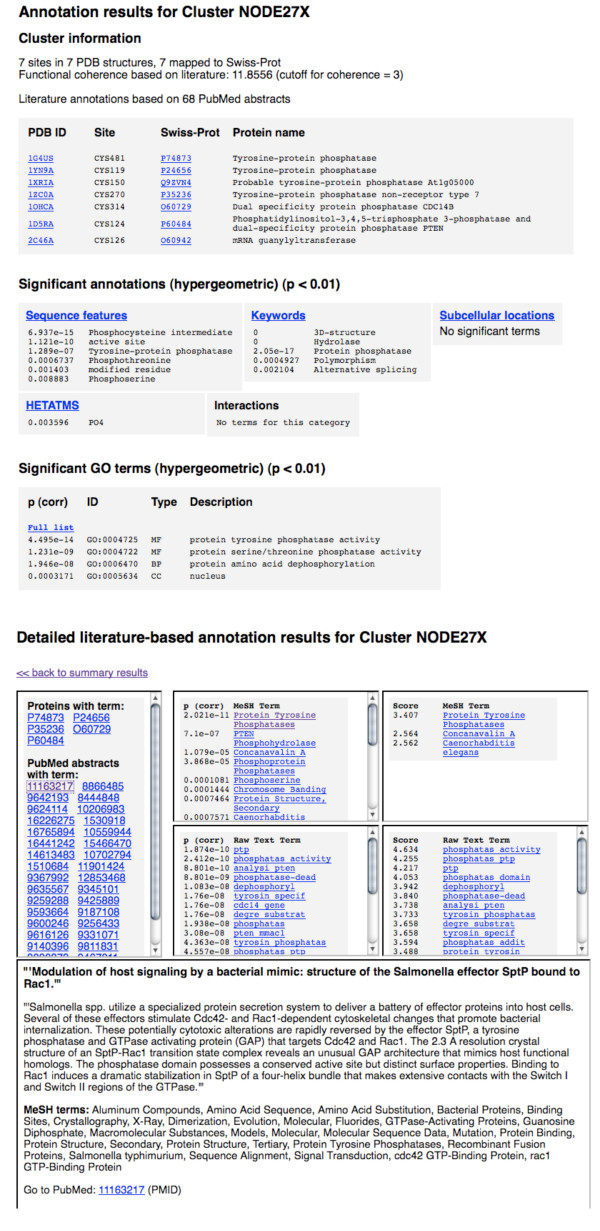
Results: In order to identify novel 3D motifs that may be associated with molecular functions, we employ an unsupervised, two-phase clustering approach that combines k-means and hierarchical clustering with knowledge-informed cluster selection and annotation methods. We applied the approach to approximately 20,000 cysteine-based protein microenvironments (3D regions 7.5 A in radius) and identified 70 interesting clusters, some of which represent known motifs (e.g. metal binding and phosphatase activity), and some of which are novel, including several zinc binding sites. Detailed annotation results are available online for all 70 clusters at http://feature.stanford.edu/clustering/cys.
Conclusions: The use of microenvironments instead of backbone geometric criteria enables flexible exploration of protein function space, and detection of recurring motifs that are discontinuous in sequence and diverse in structure. Clustering microenvironments may thus help to functionally characterize novel proteins and better understand the protein structure-function relationship.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
