

A general co-expression network-based approach to gene expression analysis: comparison and applications
- PMID: 20122284
- PMCID: PMC2829495
- DOI: 10.1186/1752-0509-4-8
A general co-expression network-based approach to gene expression analysis: comparison and applications
Abstract
Background: Co-expression network-based approaches have become popular in analyzing microarray data, such as for detecting functional gene modules. However, co-expression networks are often constructed by ad hoc methods, and network-based analyses have not been shown to outperform the conventional cluster analyses, partially due to the lack of an unbiased evaluation metric.
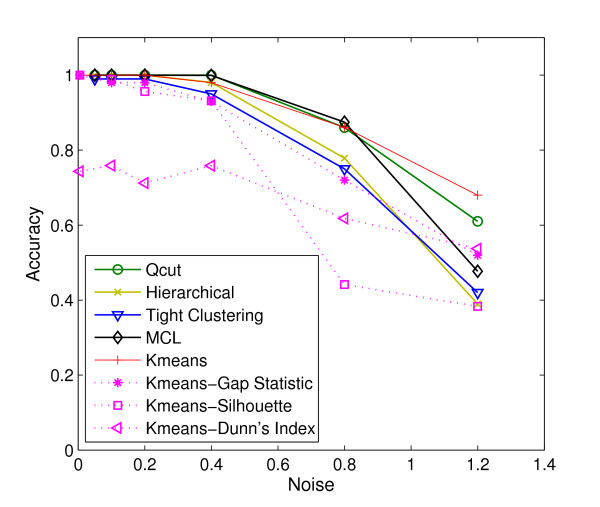
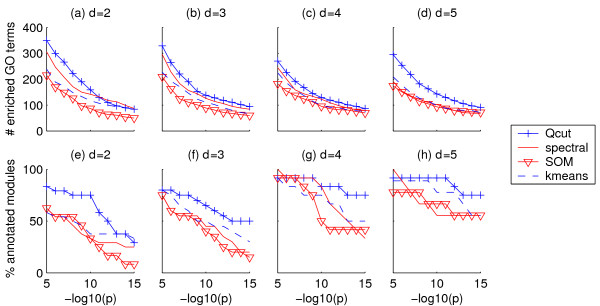
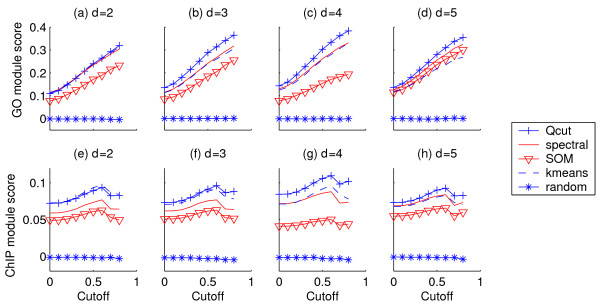
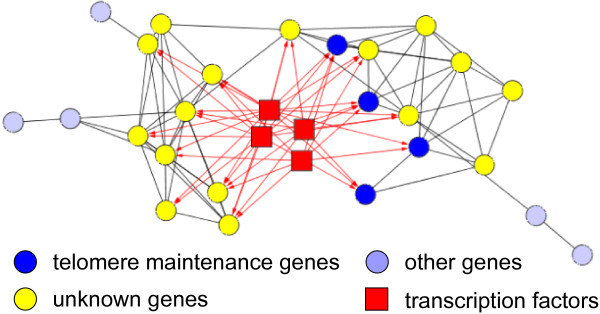
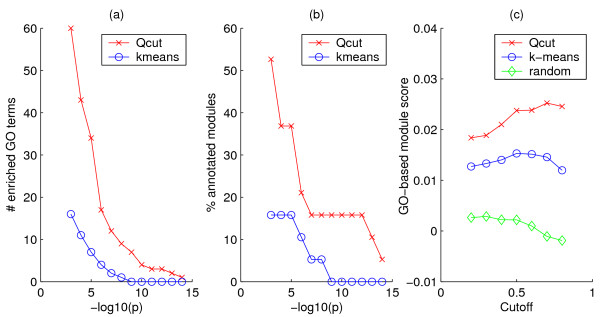
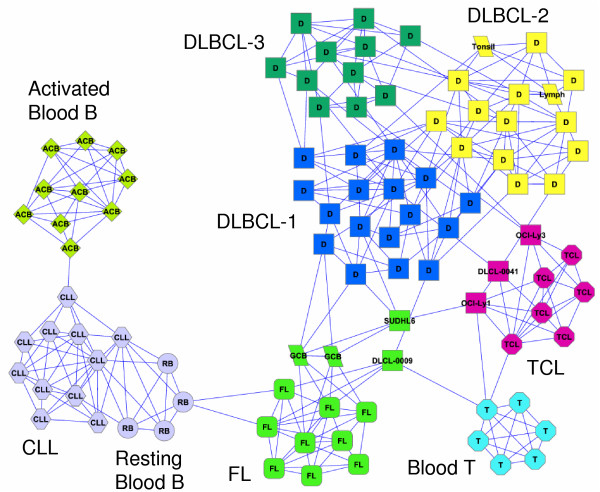
Results: Here, we develop a general co-expression network-based approach for analyzing both genes and samples in microarray data. Our approach consists of a simple but robust rank-based network construction method, a parameter-free module discovery algorithm and a novel reference network-based metric for module evaluation. We report some interesting topological properties of rank-based co-expression networks that are very different from that of value-based networks in the literature. Using a large set of synthetic and real microarray data, we demonstrate the superior performance of our approach over several popular existing algorithms. Applications of our approach to yeast, Arabidopsis and human cancer microarray data reveal many interesting modules, including a fatal subtype of lymphoma and a gene module regulating yeast telomere integrity, which were missed by the existing methods.
Conclusions: We demonstrated that our novel approach is very effective in discovering the modular structures in microarray data, both for genes and for samples. As the method is essentially parameter-free, it may be applied to large data sets where the number of clusters is difficult to estimate. The method is also very general and can be applied to other types of data. A MATLAB implementation of our algorithm can be downloaded from http://cs.utsa.edu/~jruan/Software.html.
Figures
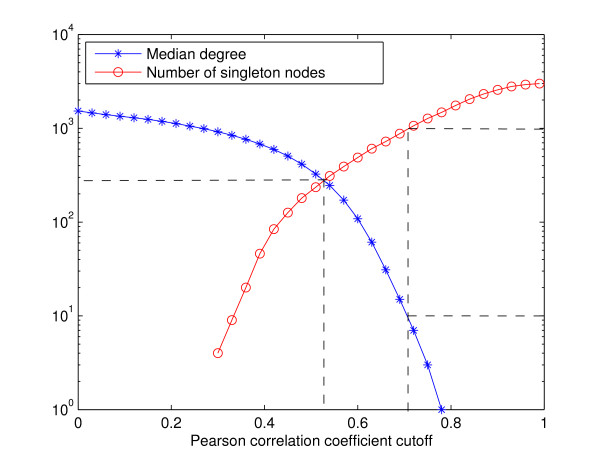
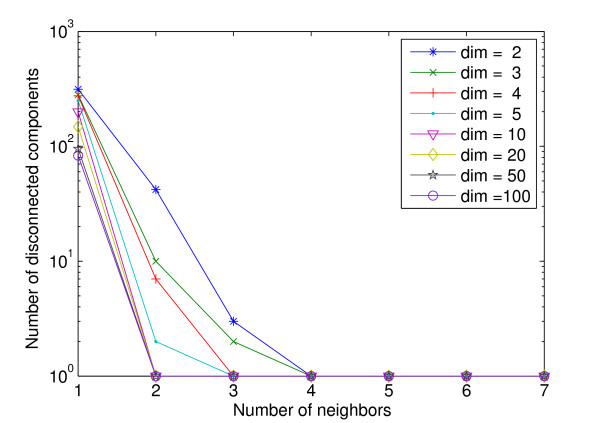
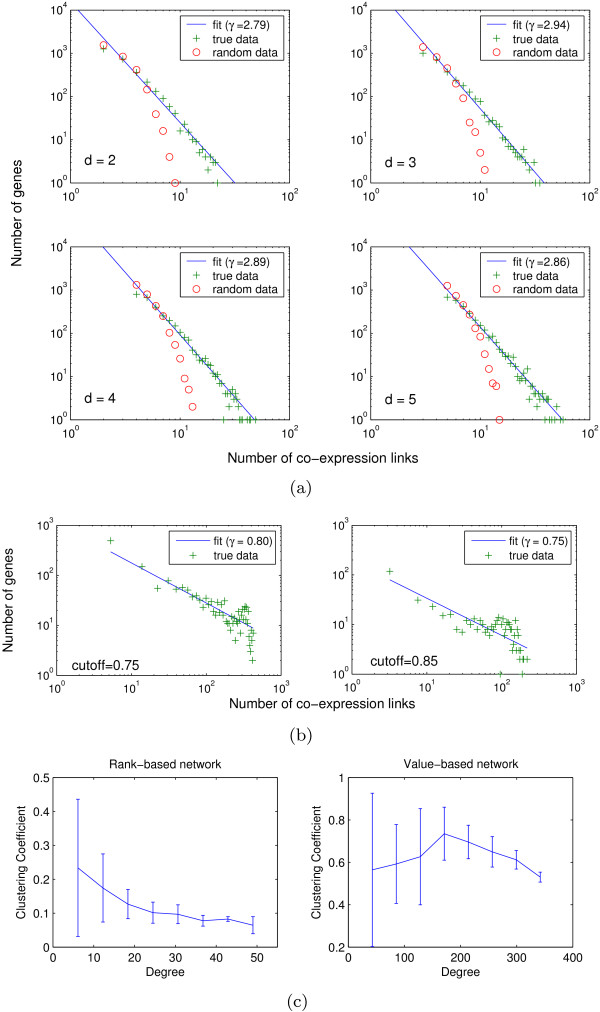
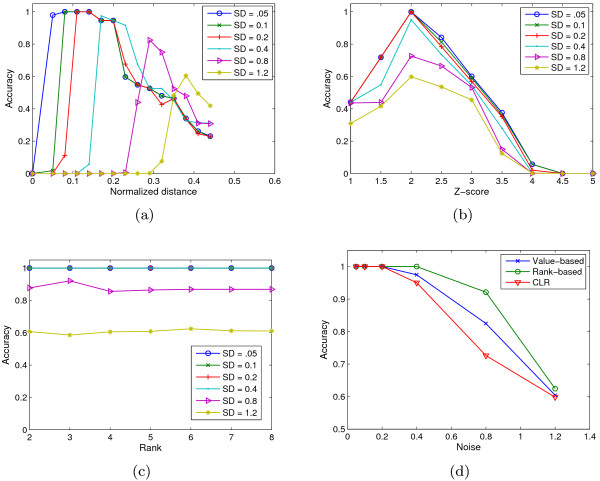
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
