

A new strategy for genome assembly using short sequence reads and reduced representation libraries
- PMID: 20123915
- PMCID: PMC2813480
- DOI: 10.1101/gr.097956.109
A new strategy for genome assembly using short sequence reads and reduced representation libraries
Abstract
We have developed a novel approach for using massively parallel short-read sequencing to generate fast and inexpensive de novo genomic assemblies comparable to those generated by capillary-based methods. The ultrashort (<100 base) sequences generated by this technology pose specific biological and computational challenges for de novo assembly of large genomes. To account for this, we devised a method for experimentally partitioning the genome using reduced representation (RR) libraries prior to assembly. We use two restriction enzymes independently to create a series of overlapping fragment libraries, each containing a tractable subset of the genome. Together, these libraries allow us to reassemble the entire genome without the need of a reference sequence. As proof of concept, we applied this approach to sequence and assembled the majority of the 125-Mb Drosophila melanogaster genome. We subsequently demonstrate the accuracy of our assembly method with meaningful comparisons against the current available D. melanogaster reference genome (dm3). The ease of assembly and accuracy for comparative genomics suggest that our approach will scale to future mammalian genome-sequencing efforts, saving both time and money without sacrificing quality.
Figures



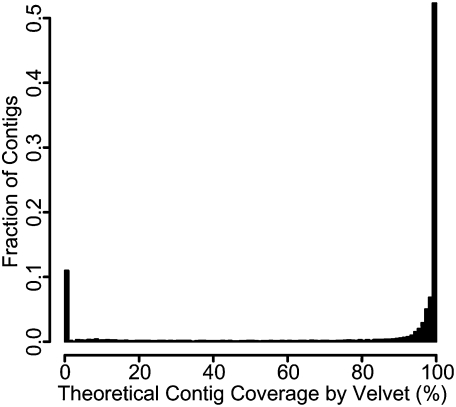





References
-
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Altshuler D, Pollara VJ, Cowles CR, Van Etten WJ, Baldwin J, Linton L, Lander ES. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous