

Imaging enzymes at work: metabolic mapping by enzyme histochemistry
- PMID: 20124092
- PMCID: PMC2874181
- DOI: 10.1369/jhc.2010.955518
Imaging enzymes at work: metabolic mapping by enzyme histochemistry
Abstract
For the understanding of functions of proteins in biological and pathological processes, reporter molecules such as fluorescent proteins have become indispensable tools for visualizing the location of these proteins in intact animals, tissues, and cells. For enzymes, imaging their activity also provides information on their function or functions, which does not necessarily correlate with their location. Metabolic mapping enables imaging of activity of enzymes. The enzyme under study forms a reaction product that is fluorescent or colored by conversion of either a fluorogenic or chromogenic substrate or a fluorescent substrate with different spectral characteristics. Most chromogenic staining methods were developed in the latter half of the twentieth century but still find new applications in modern cell biology and pathology. Fluorescence methods have rapidly evolved during the last decade. This review critically evaluates the methods that are available at present for metabolic mapping in living animals, unfixed cryostat sections of tissues, and living cells, and refers to protocols of the methods of choice.
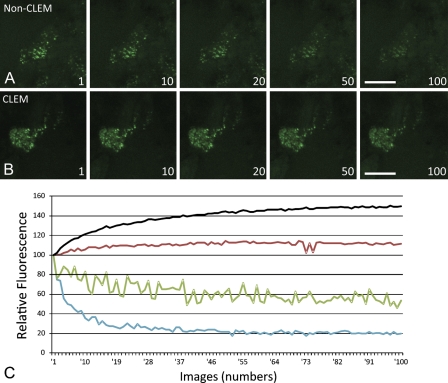
Figures








References
-
- Abu-Absi SF, Friend JR, Hansen LK, Hu W-S (2002) Structural polarity and functional bile canaliculi in rat hepatocyte spheroids. Exp Cell Res 274:56–67 - PubMed
-
- Acerenza L, Grana M (2006) On the origins of a crowded cytoplasm. J Mol Evol 63:583–590 - PubMed
-
- Akiba Y, Mizumori M, Guth PH, Engel E, Kaunitz JD (2007) Duodenal brush border intestinal alkaline phosphatase activity affects bicarbonate secretion in rats. Am J Physiol Gastrointest Liver Physiol 293:G1223–1233 - PubMed
-
- Ali AT, Penny CB, Paiker JE, Van Niekerk C, Smit A, Ferris WF, Crowther NJ (2005) Alkaline phosphatase is involved in the control of adipogenesis in the murine preadipocyte cell line, 3T3–L1. Clin Chim Acta 354:101–109 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
