

Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects
- PMID: 20124439
- PMCID: PMC2858258
- DOI: 10.1161/CIRCGENETICS.109.908905
Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects
Abstract
Background: Recent studies in mice have established that an endothelial cell protein, glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1), is essential for the lipolytic processing of triglyceride-rich lipoproteins.
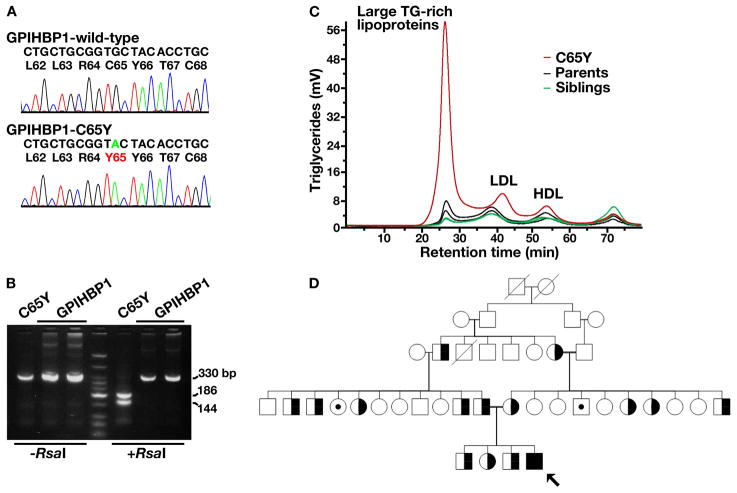
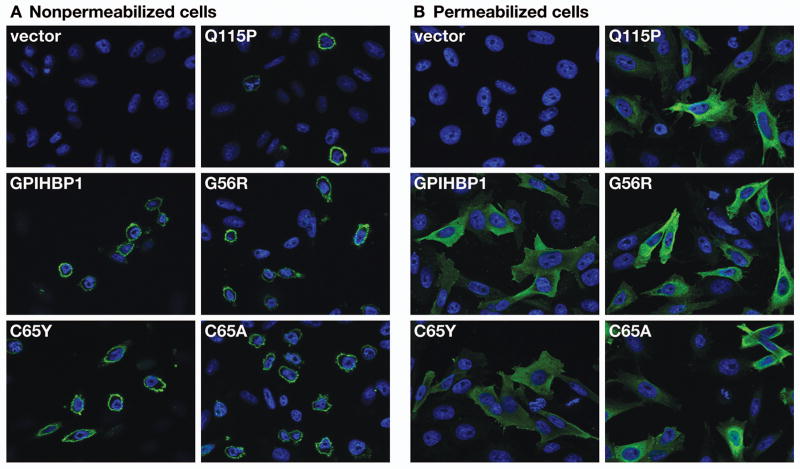
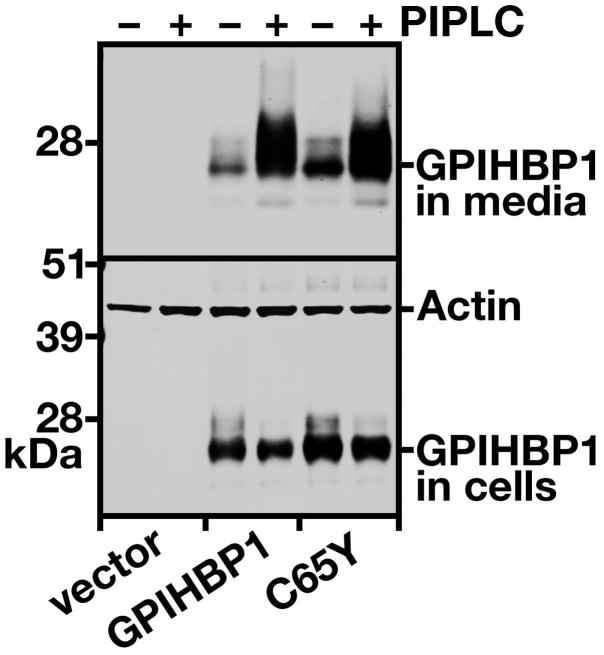
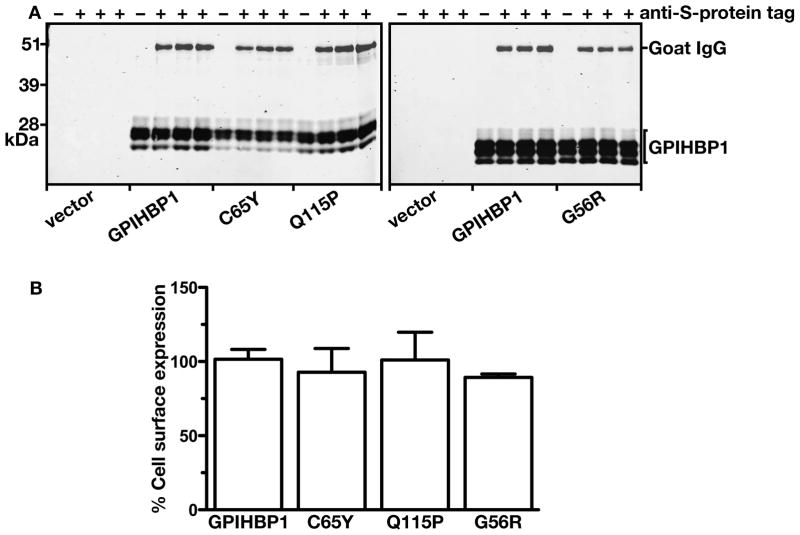
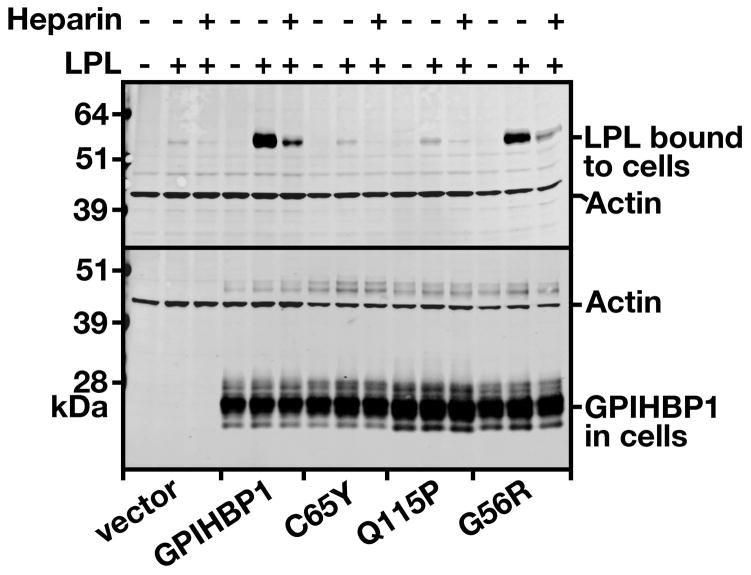
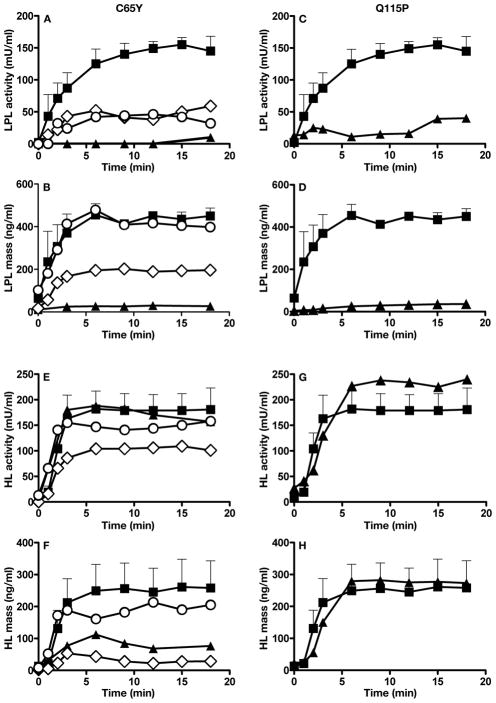
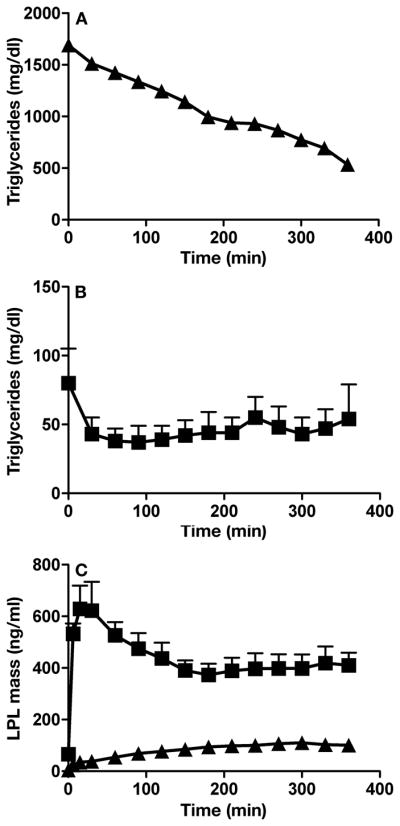
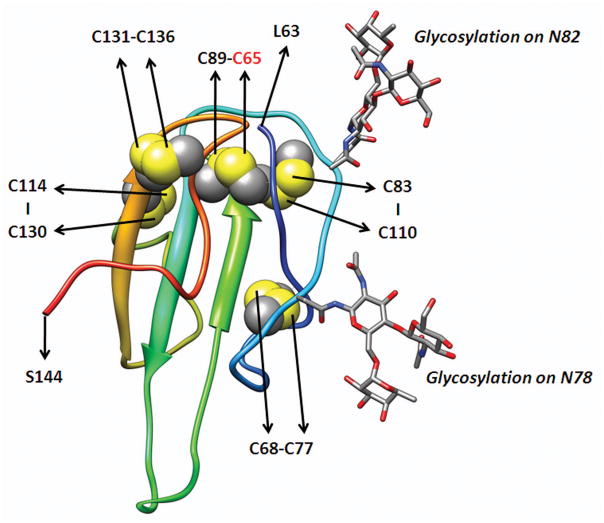
Methods and results: We report the discovery of a homozygous missense mutation in GPIHBP1 in a young boy with severe chylomicronemia. The mutation, p.C65Y, replaces a conserved cysteine in the GPIHBP1 lymphocyte antigen 6 domain with a tyrosine and is predicted to perturb protein structure by interfering with the formation of a disulfide bond. Studies with transfected Chinese hamster ovary cells showed that GPIHBP1-C65Y reaches the cell surface but has lost the ability to bind lipoprotein lipase (LPL). When the GPIHBP1-C65Y homozygote was given an intravenous bolus of heparin, only trace amounts of LPL entered the plasma. We also observed very low levels of LPL in the postheparin plasma of a subject with chylomicronemia who was homozygous for a different GPIHBP1 mutation (p.Q115P). When the GPIHBP1-Q115P homozygote was given a 6-hour infusion of heparin, a significant amount of LPL appeared in the plasma, resulting in a fall in the plasma triglyceride levels from 1780 to 120 mg/dL.
Conclusions: We identified a novel GPIHBP1 missense mutation (p.C65Y) associated with defective LPL binding in a young boy with severe chylomicronemia. We also show that homozygosity for the C65Y or Q115P mutations is associated with low levels of LPL in the postheparin plasma, demonstrating that GPIHBP1 is important for plasma triglyceride metabolism in humans.
Figures
References
-
- Wang H, Eckel RH. Lipoprotein Lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. - PubMed
-
- Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, De Sauvage FJ, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
