

Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle
- PMID: 20124730
- PMCID: PMC2827958
- DOI: 10.1172/JCI40736
Golgi and sarcolemmal neuronal NOS differentially regulate contraction-induced fatigue and vasoconstriction in exercising mouse skeletal muscle
Abstract
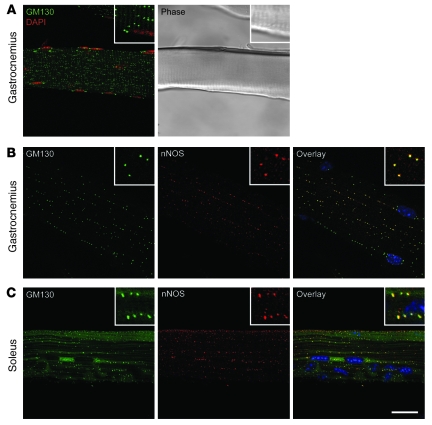
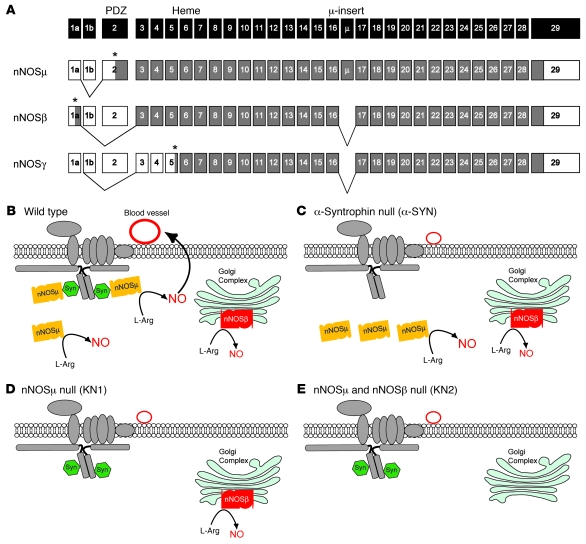
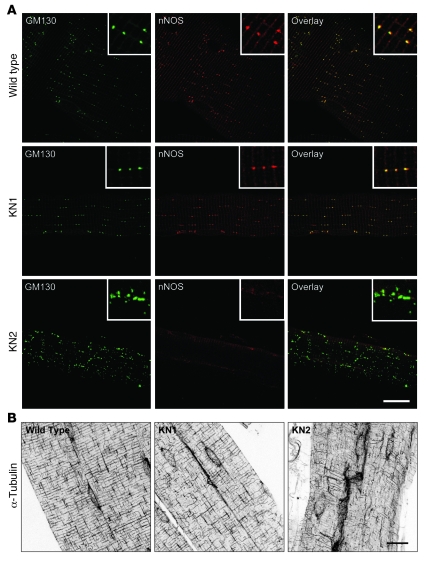
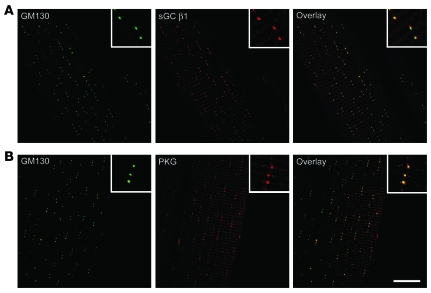
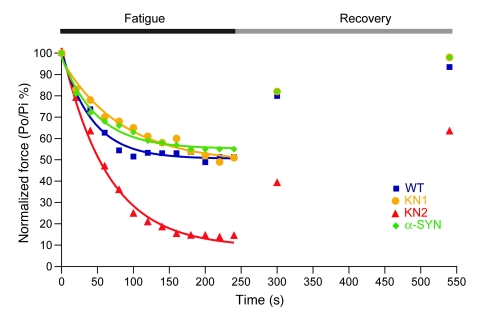
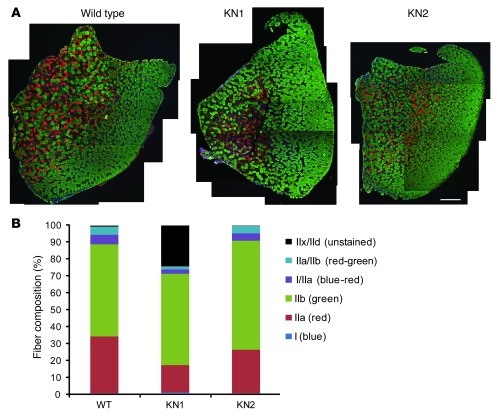
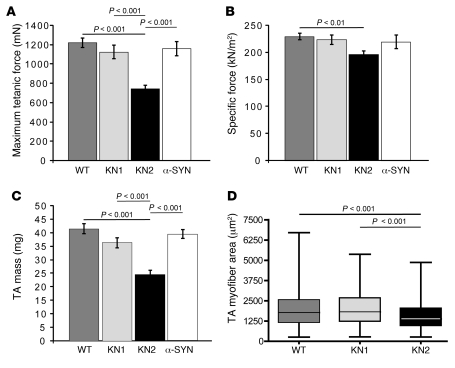
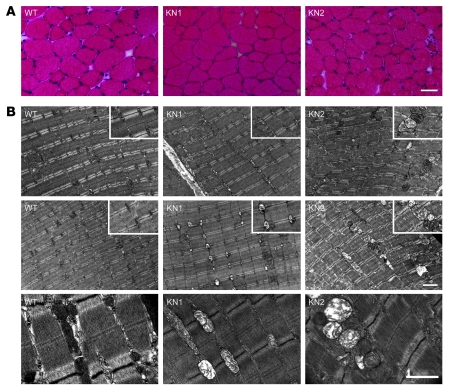
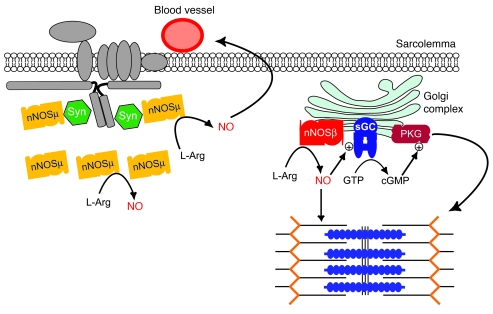
Signaling via the neuronal NOS (nNOS) splice variant nNOSmu is essential for skeletal muscle health and is commonly reduced in neuromuscular disease. nNOSmu is thought to be the predominant source of NO in skeletal muscle. Here we demonstrate the existence of what we believe to be a novel signaling pathway, mediated by the nNOS splice variant nNOSbeta, localized at the Golgi complex in mouse skeletal muscle cells. In contrast to muscles lacking nNOSmu alone, muscles missing both nNOSmu and nNOSbeta were severely myopathic, exhibiting structural defects in the microtubule cytoskeleton, Golgi complex, and mitochondria. Skeletal muscles lacking both nNOSmu and nNOSbeta were smaller in mass, intrinsically weak, highly susceptible to fatigue, and exhibited marked postexercise weakness. Our data indicate that nNOSbeta is a critical regulator of the structural and functional integrity of skeletal muscle and demonstrate the existence of 2 functionally distinct nNOS microdomains in skeletal muscle, created by the differential targeting of nNOSmu to the sarcolemma and nNOSbeta to the Golgi. We have previously shown that sarcolemmal nNOSmu matches the blood supply to the metabolic demands of active muscle. We now demonstrate that nNOSbeta simultaneously modulates the ability of skeletal muscle to maintain force production during and after exercise. We conclude therefore that nNOS splice variants are critical regulators of skeletal muscle exercise performance.
Figures
References
-
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81(1):209–237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
