

A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint
- PMID: 20126263
- PMCID: PMC2811157
- DOI: 10.1371/journal.pbio.1000287
A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint
Abstract
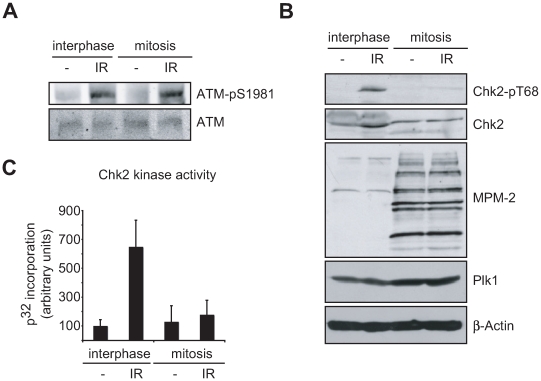
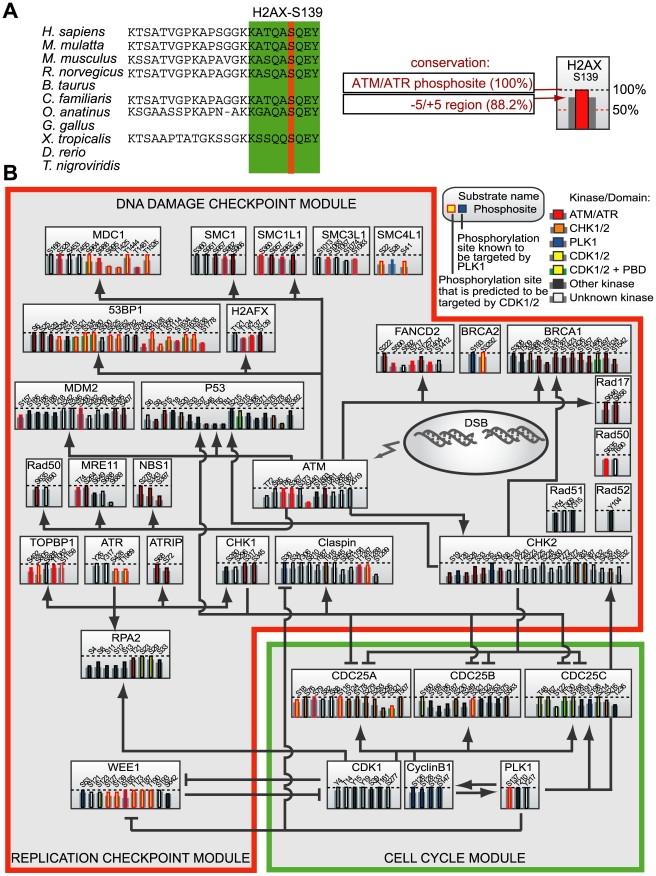
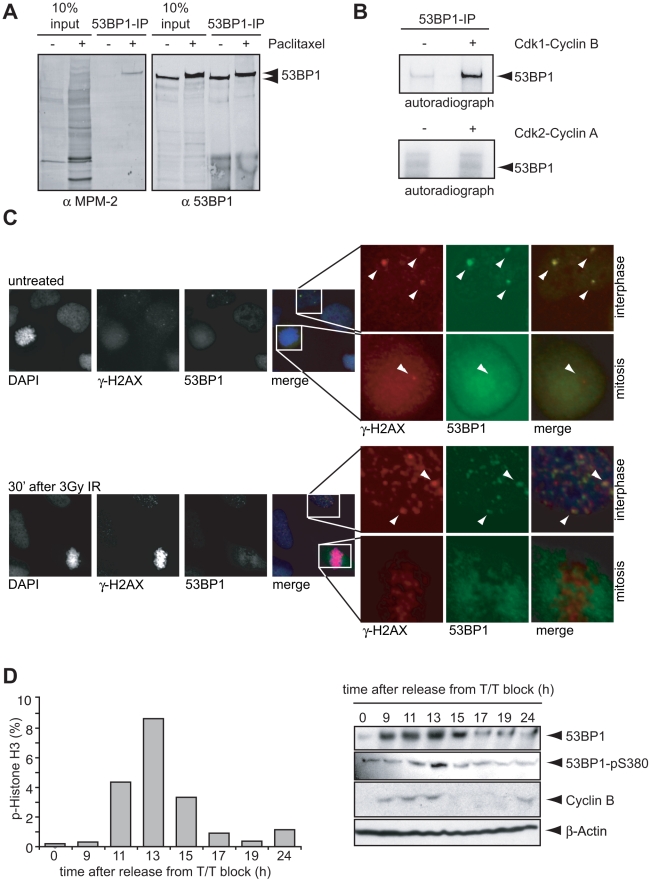
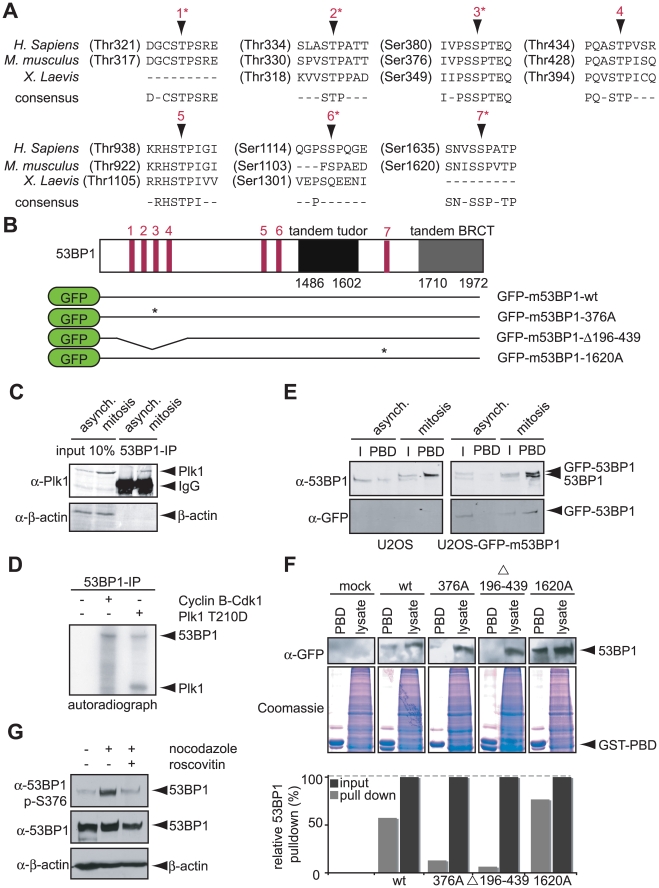
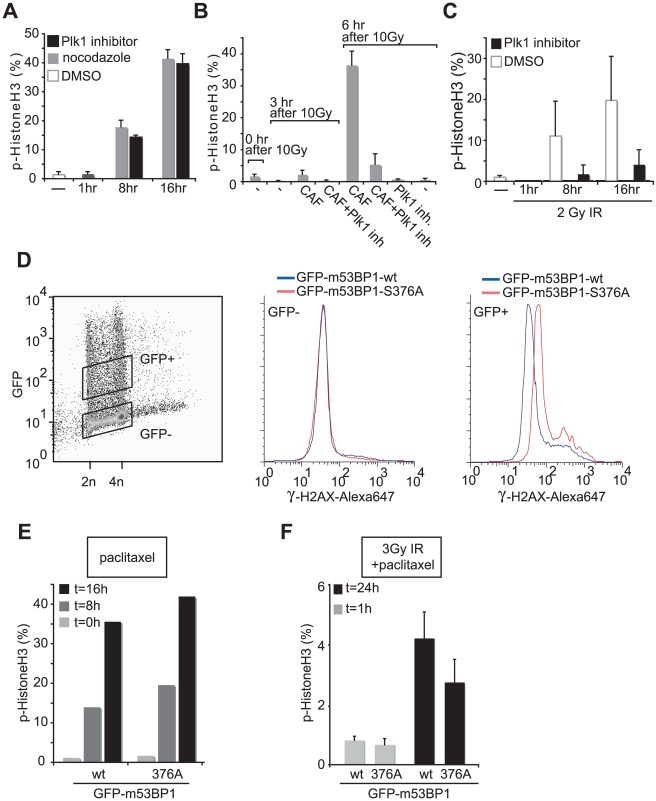
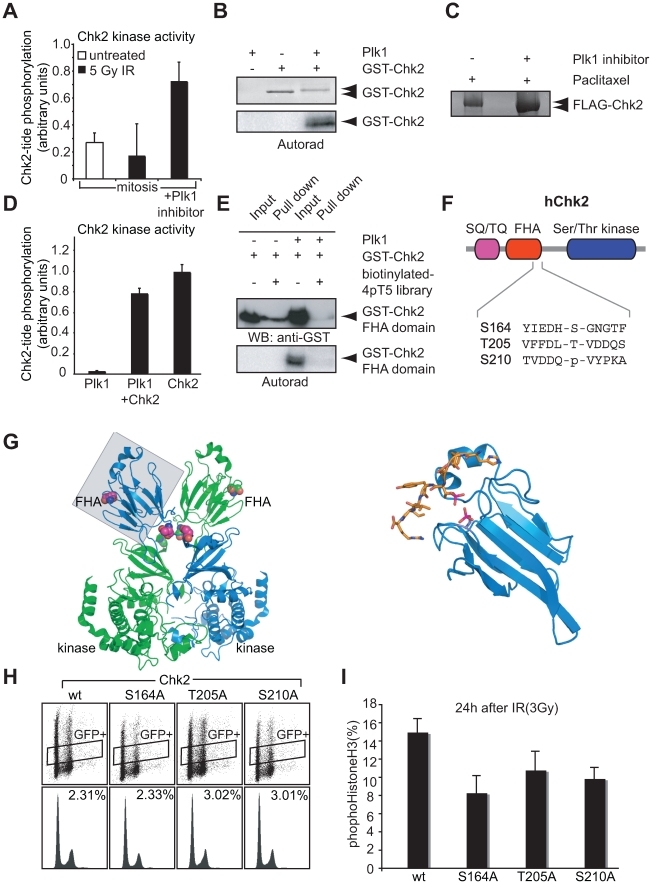
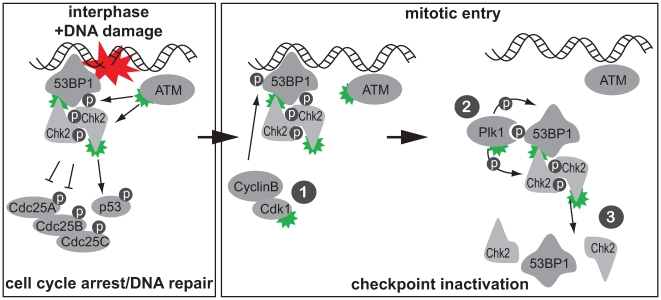
DNA damage checkpoints arrest cell cycle progression to facilitate DNA repair. The ability to survive genotoxic insults depends not only on the initiation of cell cycle checkpoints but also on checkpoint maintenance. While activation of DNA damage checkpoints has been studied extensively, molecular mechanisms involved in sustaining and ultimately inactivating cell cycle checkpoints are largely unknown. Here, we explored feedback mechanisms that control the maintenance and termination of checkpoint function by computationally identifying an evolutionary conserved mitotic phosphorylation network within the DNA damage response. We demonstrate that the non-enzymatic checkpoint adaptor protein 53BP1 is an in vivo target of the cell cycle kinases Cyclin-dependent kinase-1 and Polo-like kinase-1 (Plk1). We show that Plk1 binds 53BP1 during mitosis and that this interaction is required for proper inactivation of the DNA damage checkpoint. 53BP1 mutants that are unable to bind Plk1 fail to restart the cell cycle after ionizing radiation-mediated cell cycle arrest. Importantly, we show that Plk1 also phosphorylates the 53BP1-binding checkpoint kinase Chk2 to inactivate its FHA domain and inhibit its kinase activity in mammalian cells. Thus, a mitotic kinase-mediated negative feedback loop regulates the ATM-Chk2 branch of the DNA damage signaling network by phosphorylating conserved sites in 53BP1 and Chk2 to inactivate checkpoint signaling and control checkpoint duration.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Polo-like kinase 1 inhibits DNA damage response during mitosis.Cell Cycle. 2015;14(2):219-31. doi: 10.4161/15384101.2014.977067. Cell Cycle. 2015. PMID: 25607646 Free PMC article.
-
Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage.Cell Cycle. 2005 Jan;4(1):166-71. doi: 10.4161/cc.4.1.1348. Epub 2005 Jan 5. Cell Cycle. 2005. PMID: 15611664
-
FRET-Based Sorting of Live Cells Reveals Shifted Balance between PLK1 and CDK1 Activities During Checkpoint Recovery.Cells. 2020 Sep 19;9(9):2126. doi: 10.3390/cells9092126. Cells. 2020. PMID: 32961751 Free PMC article.
-
Cell cycle re-entry mechanisms after DNA damage checkpoints: giving it some gas to shut off the breaks!Cell Cycle. 2010 Jun 1;9(11):2097-101. doi: 10.4161/cc.9.11.11840. Epub 2010 Jun 1. Cell Cycle. 2010. PMID: 20505366 Review.
-
Polo-Like Kinase 1 and DNA Damage Response.DNA Cell Biol. 2024 Sep;43(9):430-437. doi: 10.1089/dna.2024.0018. Epub 2024 Jul 3. DNA Cell Biol. 2024. PMID: 38959179 Review.
Cited by
-
Vaccinia-related kinase 1 (VRK1) is an upstream nucleosomal kinase required for the assembly of 53BP1 foci in response to ionizing radiation-induced DNA damage.J Biol Chem. 2012 Jul 6;287(28):23757-68. doi: 10.1074/jbc.M112.353102. Epub 2012 May 22. J Biol Chem. 2012. PMID: 22621922 Free PMC article.
-
Correlative Multi-Modal Microscopy: A Novel Pipeline for Optimizing Fluorescence Microscopy Resolutions in Biological Applications.Cells. 2023 Jan 17;12(3):354. doi: 10.3390/cells12030354. Cells. 2023. PMID: 36766696 Free PMC article.
-
PLK1 promotes the mitotic surveillance pathway by controlling cytosolic 53BP1 availability.EMBO Rep. 2023 Dec 6;24(12):e57234. doi: 10.15252/embr.202357234. Epub 2023 Oct 27. EMBO Rep. 2023. PMID: 37888778 Free PMC article.
-
DNA damage response is suppressed by the high cyclin-dependent kinase 1 activity in mitotic mammalian cells.J Biol Chem. 2011 Oct 14;286(41):35899-35905. doi: 10.1074/jbc.M111.267690. Epub 2011 Aug 30. J Biol Chem. 2011. PMID: 21878640 Free PMC article.
-
Perturbation biology: inferring signaling networks in cellular systems.PLoS Comput Biol. 2013;9(12):e1003290. doi: 10.1371/journal.pcbi.1003290. Epub 2013 Dec 19. PLoS Comput Biol. 2013. PMID: 24367245 Free PMC article.
References
-
- Hoeijmakers J. H. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. - PubMed
-
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol. 2007;19:238–245. - PubMed
-
- Harper J. W, Elledge S. J. The DNA damage response: ten years after. Mol Cell. 2007;28:739–745. - PubMed
-
- Kastan M. B, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. - PubMed
-
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
