

Whole-genome sequencing of a laboratory-evolved yeast strain
- PMID: 20128923
- PMCID: PMC2829512
- DOI: 10.1186/1471-2164-11-88
Whole-genome sequencing of a laboratory-evolved yeast strain
Abstract
Background: Experimental evolution of microbial populations provides a unique opportunity to study evolutionary adaptation in response to controlled selective pressures. However, until recently it has been difficult to identify the precise genetic changes underlying adaptation at a genome-wide scale. New DNA sequencing technologies now allow the genome of parental and evolved strains of microorganisms to be rapidly determined.
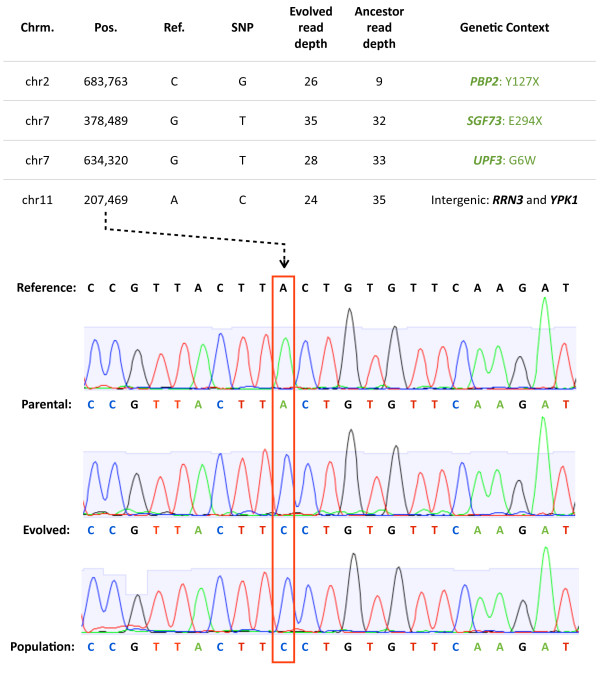
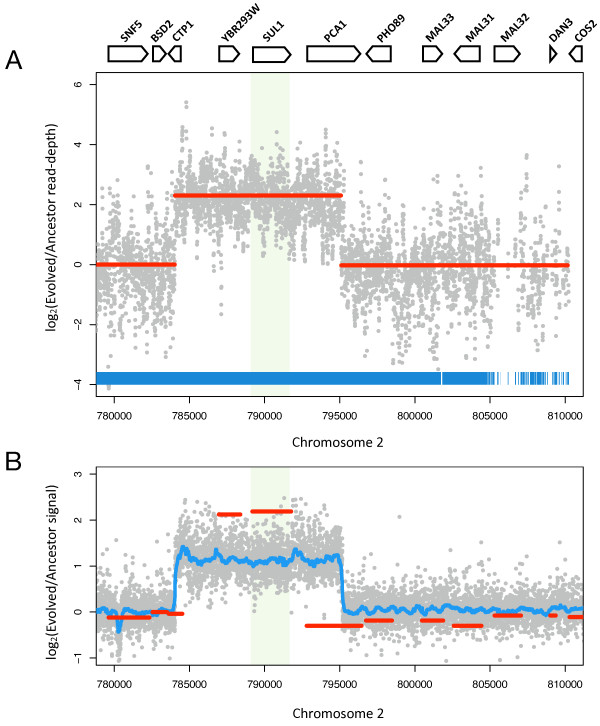
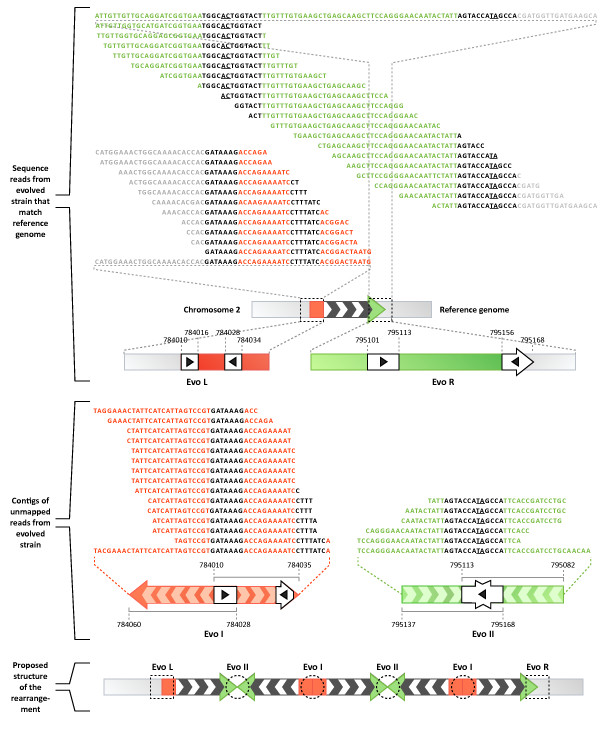
Results: We sequenced >93.5% of the genome of a laboratory-evolved strain of the yeast Saccharomyces cerevisiae and its ancestor at >28x depth. Both single nucleotide polymorphisms and copy number amplifications were found, with specific gains over array-based methodologies previously used to analyze these genomes. Applying a segmentation algorithm to quantify structural changes, we determined the approximate genomic boundaries of a 5x gene amplification. These boundaries guided the recovery of breakpoint sequences, which provide insights into the nature of a complex genomic rearrangement.
Conclusions: This study suggests that whole-genome sequencing can provide a rapid approach to uncover the genetic basis of evolutionary adaptations, with further applications in the study of laboratory selections and mutagenesis screens. In addition, we show how single-end, short read sequencing data can provide detailed information about structural rearrangements, and generate predictions about the genomic features and processes that underlie genome plasticity.
Figures
References
-
- Herring C, Raghunathan A, Honisch C, Patel T, Applebee M, Joyce A, Albert T, Blattner F, Boom D Van Den, Cantor C, Palsson B. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
