

Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase
- PMID: 20129920
- PMCID: PMC2856272
- DOI: 10.1074/jbc.M110.108688
Activation of AMP-activated protein kinase by vascular endothelial growth factor mediates endothelial angiogenesis independently of nitric-oxide synthase
Abstract
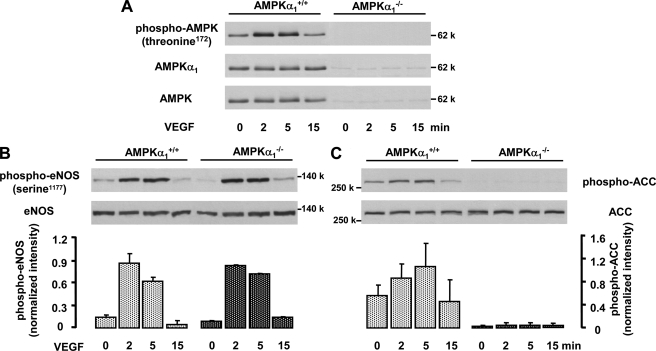
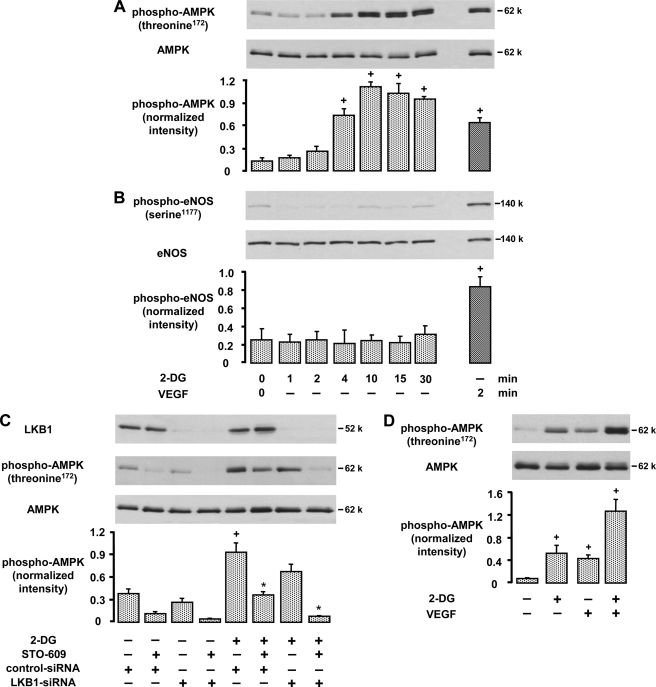
AMP-activated protein kinase (AMPK) is a sensor of cellular energy state and a regulator of cellular homeostasis. In endothelial cells, AMPK is stimulated via the upstream kinases LKB1 and Ca(2+)/calmodulin-dependent protein kinase kinase beta (CaMKKbeta). Previously, AMPK has been reported to activate endothelial nitric-oxide synthase (eNOS). Using genetic and pharmacological approaches, we show that vascular endothelial growth factor (VEGF) stimulates AMPK in human and mice endothelial cells via CaMKKbeta. VEGF-induced AMPK activation is potentiated under conditions of energy deprivation induced by 2-deoxyglucose. To investigate the role of AMPK in endothelial function, CaMKKbeta, AMPKalpha1, or AMPKalpha2 was down-regulated by RNA interference, and studies in AMPKalpha1(-/-) mice were performed. We demonstrate that AMPK does not mediate eNOS phosphorylation at serine residue 1177 or 633, NO- dependent cGMP generation, or Akt phosphorylation in response to VEGF. Using inhibitors of eNOS or soluble guanylyl cyclase and small interfering RNA against eNOS, we show that NO does not act upstream of AMPK. Taken together, these data indicate that VEGF-stimulated AMPK and eNOS pathways act independently of each other. However, acetyl-CoA carboxylase, a key enzyme in the regulation of fatty acid oxidation, was phosphorylated in response to VEGF in an AMPKalpha1- and AMPKalpha2-dependent manner. Our results show that AMPKalpha1 plays an essential role in VEGF-induced angiogenesis in vitro (tube formation and sprouting from spheroids) and in vivo (Matrigel plug assay). In contrast, AMPKalpha2 was not involved in VEGF-triggered sprouting. The data suggest that AMPKalpha1 promotes VEGF-induced angiogenesis independently of eNOS, possibly by providing energy via inhibition of acetyl-CoA carboxylase.
Figures







Similar articles
-
eNOS activation mediated by AMPK after stimulation of endothelial cells with histamine or thrombin is dependent on LKB1.Biochim Biophys Acta. 2011 Feb;1813(2):322-31. doi: 10.1016/j.bbamcr.2010.12.001. Epub 2010 Dec 9. Biochim Biophys Acta. 2011. PMID: 21145922
-
Rutaecarpine Increases Nitric Oxide Synthesis via eNOS Phosphorylation by TRPV1-Dependent CaMKII and CaMKKβ/AMPK Signaling Pathway in Human Endothelial Cells.Int J Mol Sci. 2021 Aug 30;22(17):9407. doi: 10.3390/ijms22179407. Int J Mol Sci. 2021. PMID: 34502308 Free PMC article.
-
Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway.J Biol Chem. 2007 Jul 13;282(28):20351-64. doi: 10.1074/jbc.M702182200. Epub 2007 May 22. J Biol Chem. 2007. PMID: 17519230
-
Inositol polyphosphate multikinase: an emerging player for the central action of AMP-activated protein kinase.Biochem Biophys Res Commun. 2012 Apr 27;421(1):1-3. doi: 10.1016/j.bbrc.2012.04.010. Epub 2012 Apr 7. Biochem Biophys Res Commun. 2012. PMID: 22503973 Free PMC article. Review.
-
AMP-Activated Protein Kinase: Do We Need Activators or Inhibitors to Treat or Prevent Cancer?Int J Mol Sci. 2020 Dec 27;22(1):186. doi: 10.3390/ijms22010186. Int J Mol Sci. 2020. PMID: 33375416 Free PMC article. Review.
Cited by
-
Influence of sphingosine-1-phosphate signaling on HCMV replication in human embryonal lung fibroblasts.Med Microbiol Immunol. 2018 Aug;207(3-4):227-242. doi: 10.1007/s00430-018-0543-4. Epub 2018 Apr 26. Med Microbiol Immunol. 2018. PMID: 29700602
-
Endothelial cell CD36 deficiency prevents normal angiogenesis and vascular repair.Am J Transl Res. 2020 Dec 15;12(12):7737-7761. eCollection 2020. Am J Transl Res. 2020. PMID: 33437358 Free PMC article.
-
The Association Between Inflammatory and Oxidative Stress Biomarkers and Plasma Metabolites in a Longitudinal Study of Healthy Male Welders.J Inflamm Res. 2021 Jun 29;14:2825-2839. doi: 10.2147/JIR.S316262. eCollection 2021. J Inflamm Res. 2021. PMID: 34234508 Free PMC article.
-
Keeping the home fires burning: AMP-activated protein kinase.J R Soc Interface. 2018 Jan;15(138):20170774. doi: 10.1098/rsif.2017.0774. J R Soc Interface. 2018. PMID: 29343628 Free PMC article. Review.
-
The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? †.Open Biol. 2019 Jul 26;9(7):190099. doi: 10.1098/rsob.190099. Epub 2019 Jul 10. Open Biol. 2019. PMID: 31288625 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
