

LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance
- PMID: 20130076
- PMCID: PMC2853996
- DOI: 10.1210/jc.2009-1293
LMNA mutations, skeletal muscle lipid metabolism, and insulin resistance
Erratum in
- J Clin Endocrinol Metab. 2010 May;95(5):2521
Abstract
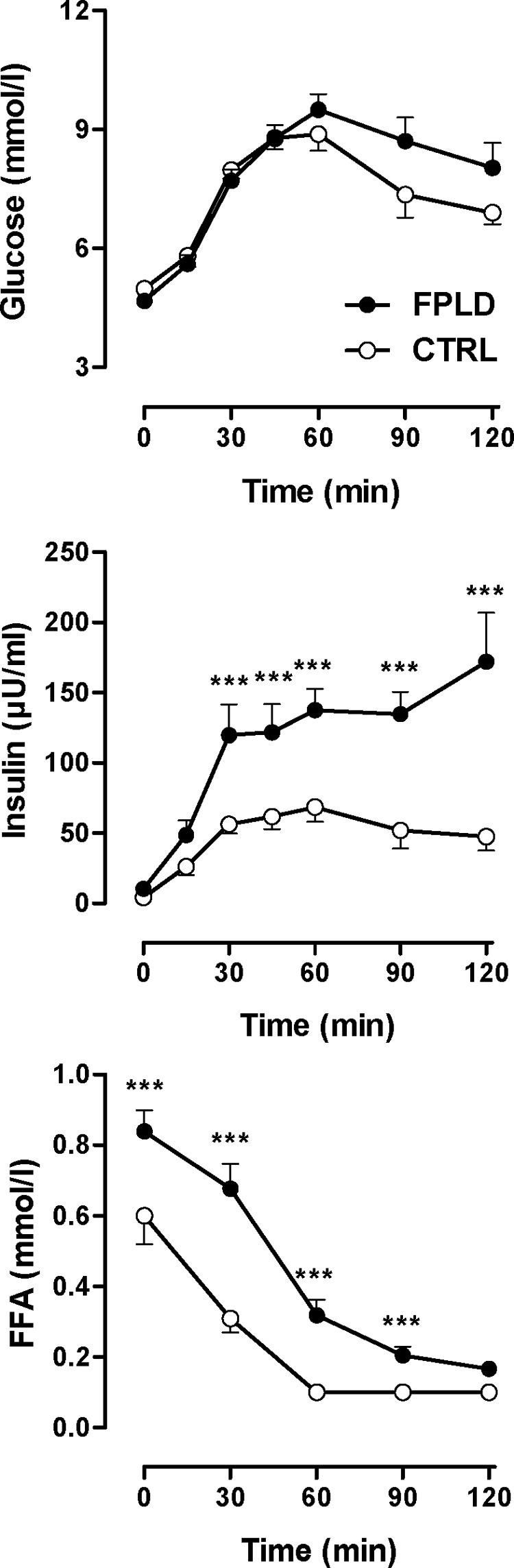
Context: Type 2 familial partial lipodystrophy (FPLD) is an autosomal-dominant lamin A/C-related disease associated with exercise intolerance, muscular pain, and insulin resistance. The symptoms may all be explained by defective metabolism; however, metabolism at the tissue level has not been investigated.
Objective: We hypothesized that in FPLD, insulin resistance and impaired aerobic exercise capacity are explained by a common underlying mechanism, presumably a muscular metabolic defect.
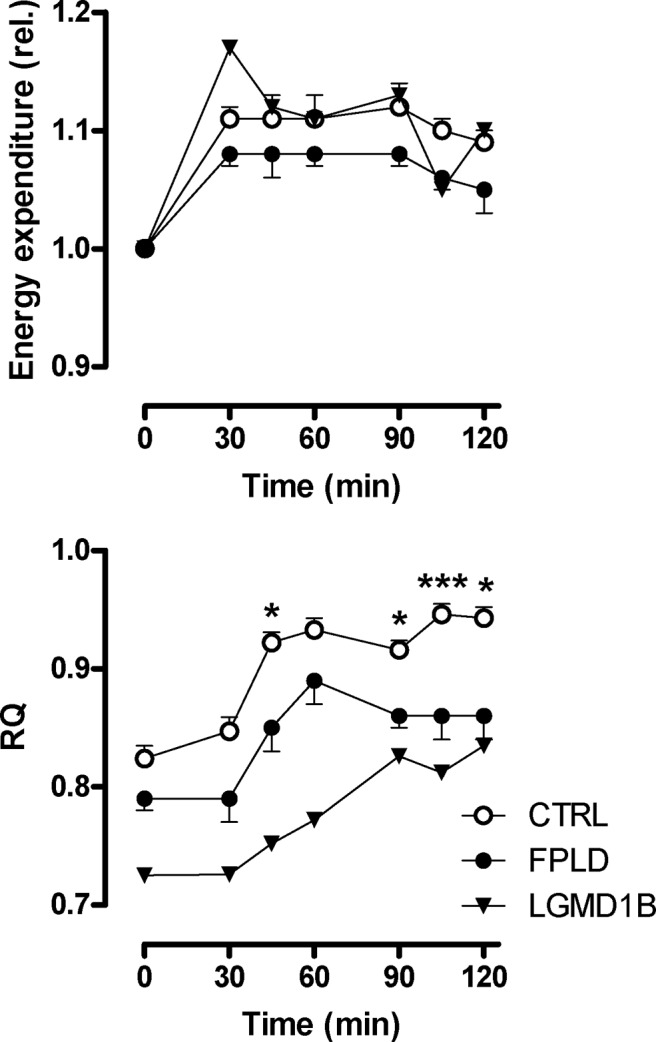
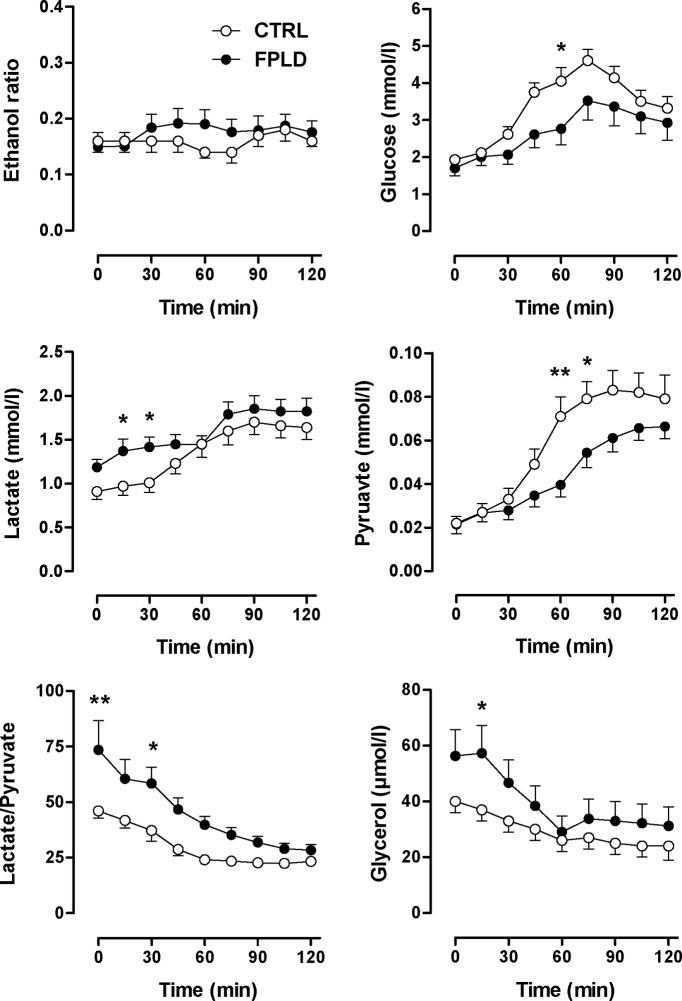
Patients and methods: Carbohydrate and lipid metabolism was studied on 10 FPLD patients, one patient with limb-girdle muscular dystrophy (LGMD1B, a different lamin A/C disease), and 10 healthy control subjects before and during an oral glucose tolerance test by indirect calorimetry and im microdialysis. Muscle biopsies were taken for in vitro studies.
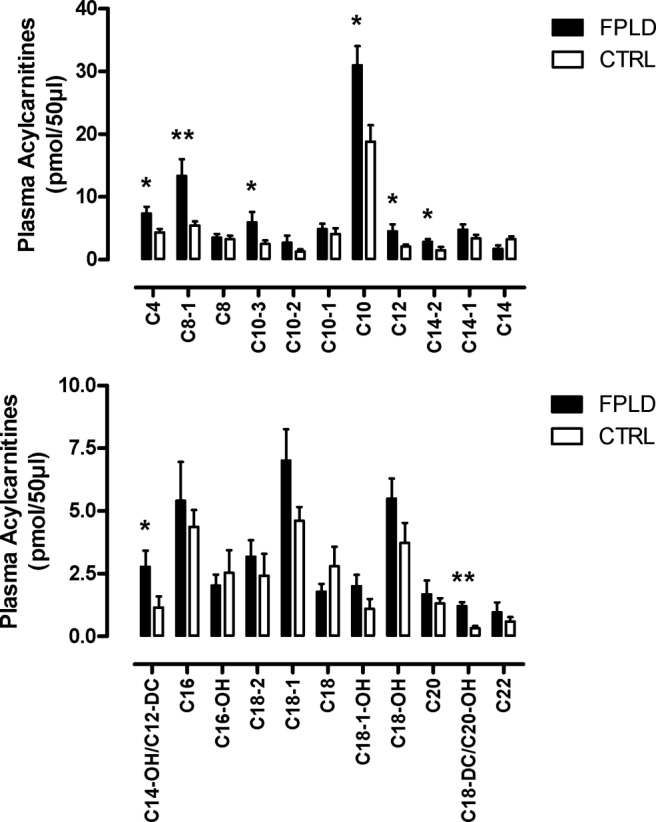
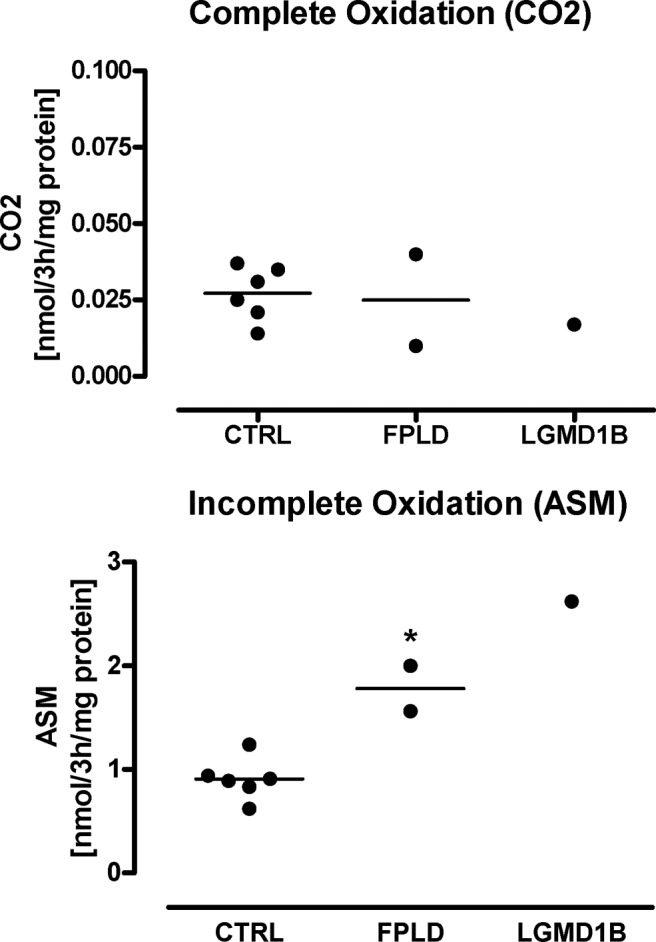
Results: We observed marked increased skeletal muscle fatty acid beta-oxidation rate in vitro and in vivo, even after glucose ingestion in FPLD patients. However, fatty acid oxidation was largely incomplete and accompanied by increased ketogenesis. The lipid oxidation abnormality was associated with impaired glucose disposition through reduction in glucose oxidation, rather than decreased cellular glucose uptake. A microarray showed down-regulation of complex I respiratory chain, glycolysis, and nuclear transport genes. Although not overtly insulin resistant, the LGMD1B patient showed similar metabolic derangements as the FPLD patients.
Conclusions: Our study suggests imbalance between lipid oxidation and oxidative glucose metabolism in FPLD and LGMD1B patients. The observation suggests an intrinsic defect in skeletal muscle metabolism due to lamin A/C dysfunction. The metabolic FPLD phenotype likely results from this intrinsic defect combined with lipodystrophic "lipid pressure" due to decreased adipose tissue lipid storage capacity.
Figures
References
-
- Cao H, Hegele RA 2000 Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy. Hum Mol Genet 9:109–112 - PubMed
-
- Speckman RA, Garg A, Du F, Bennett L, Veile R, Arioglu E, Taylor SI, Lovett M, Bowcock AM 2000 Mutational and haplotype analyses of families with familial partial lipodystrophy (Dunnigan variety) reveal recurrent missense mutations in the globular C-terminal domain of lamin A/C. Am J Hum Genet 66:1192–1198 - PMC - PubMed
-
- Shackleton S, Lloyd DJ, Jackson SN, Evans R, Niermeijer MF, Singh BM, Schmidt H, Brabant G, Kumar S, Durrington PN, Gregory S, O'Rahilly S, Trembath RC 2000 LMNA, encoding lamin A/C, is mutated in partial lipodystrophy. Nat Genet 24:153–156 - PubMed
-
- Hegele RA, Cao H, Anderson CM, Hramiak IM 2000 Heterogeneity of nuclear lamin A mutations in Dunnigan-type familial partial lipodystrophy. J Clin Endocrinol Metab 85:3431–3435 - PubMed
-
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, Wilson KL 2005 The nuclear lamina comes of age. Nat Rev Mol Cell Biol 6:21–31 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
