

Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice
- PMID: 20131403
- PMCID: PMC2930014
- DOI: 10.1002/hep.23511
Mammalian target of rapamycin regulates vascular endothelial growth factor-dependent liver cyst growth in polycystin-2-defective mice
Abstract
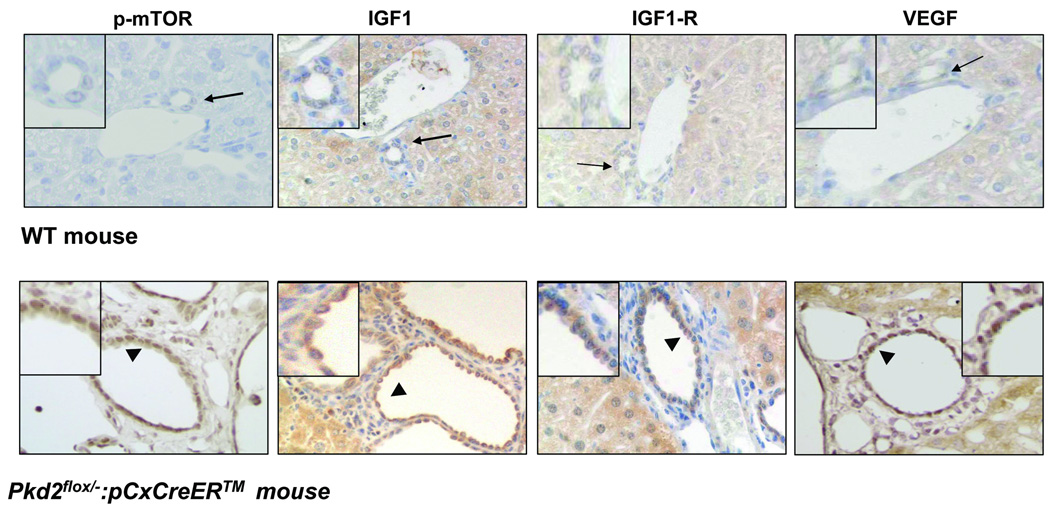
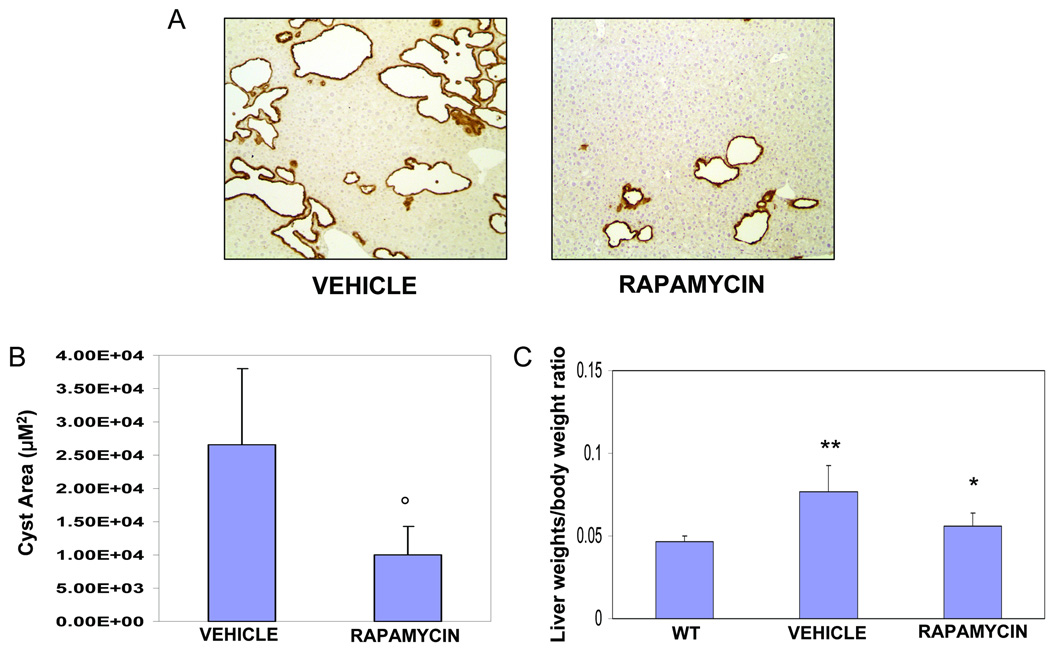
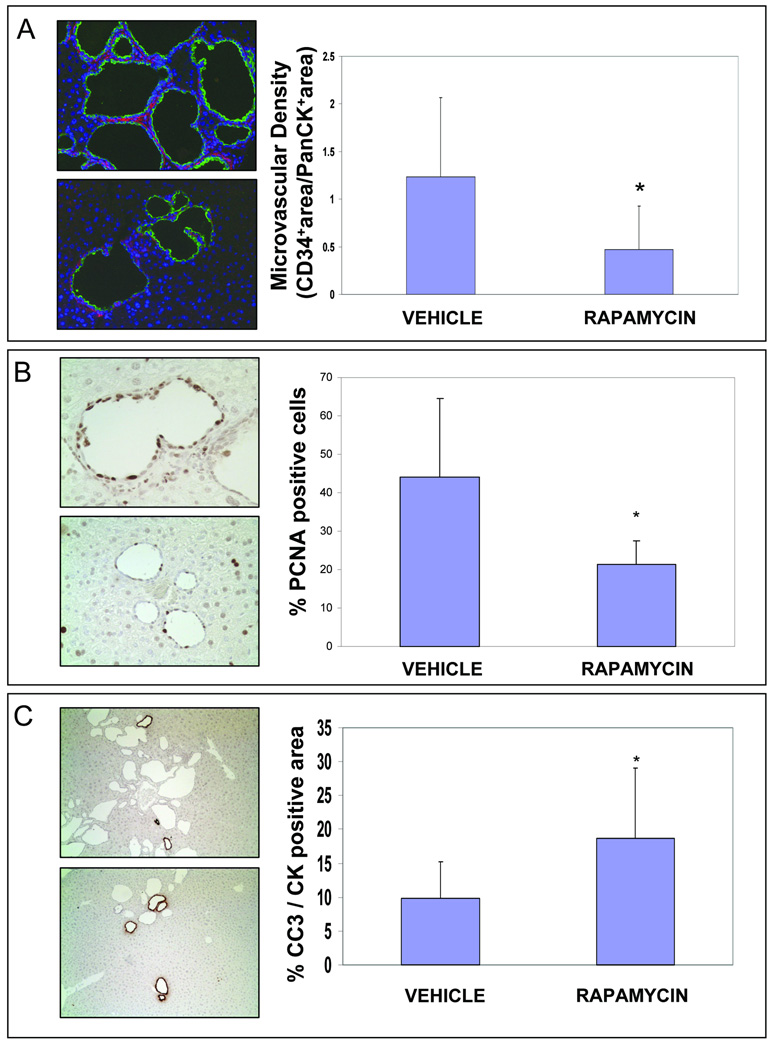
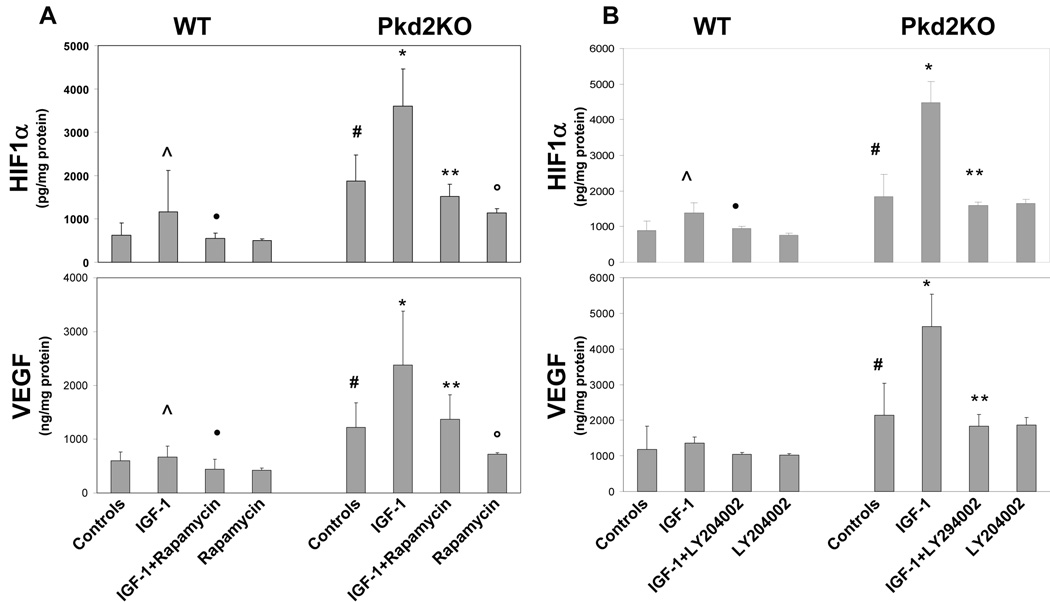
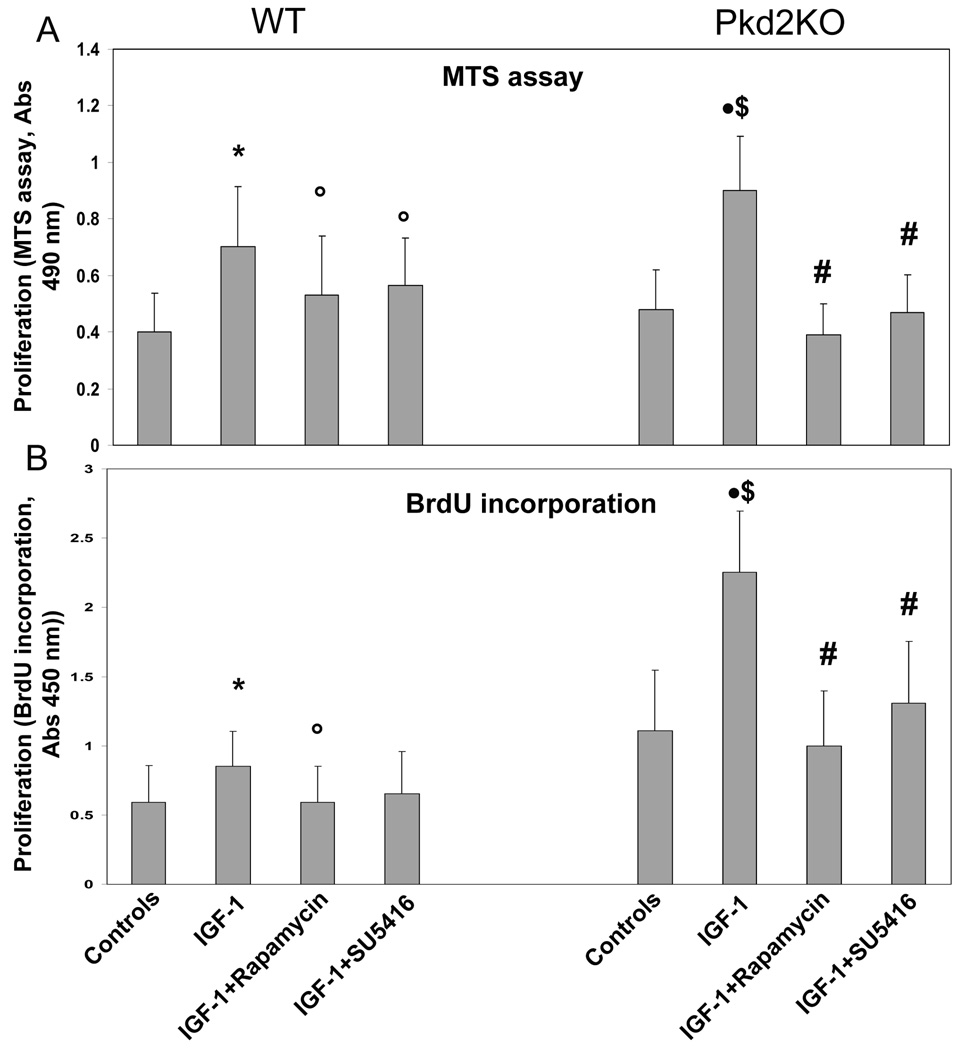
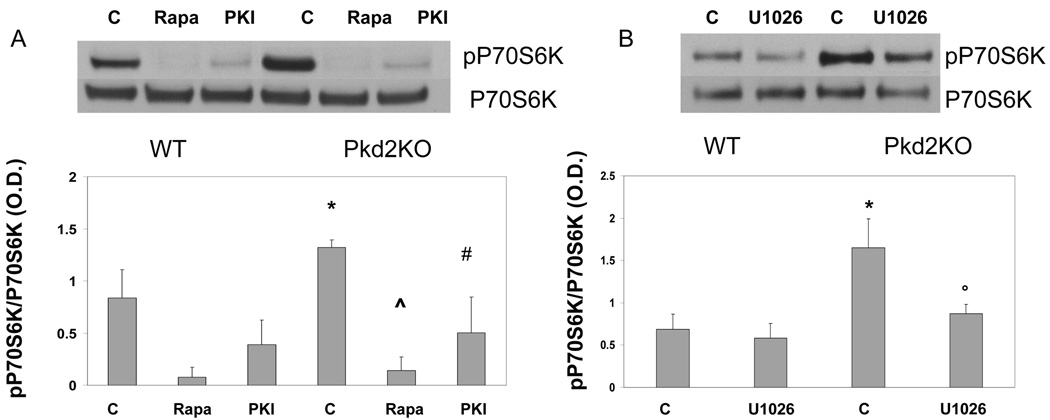
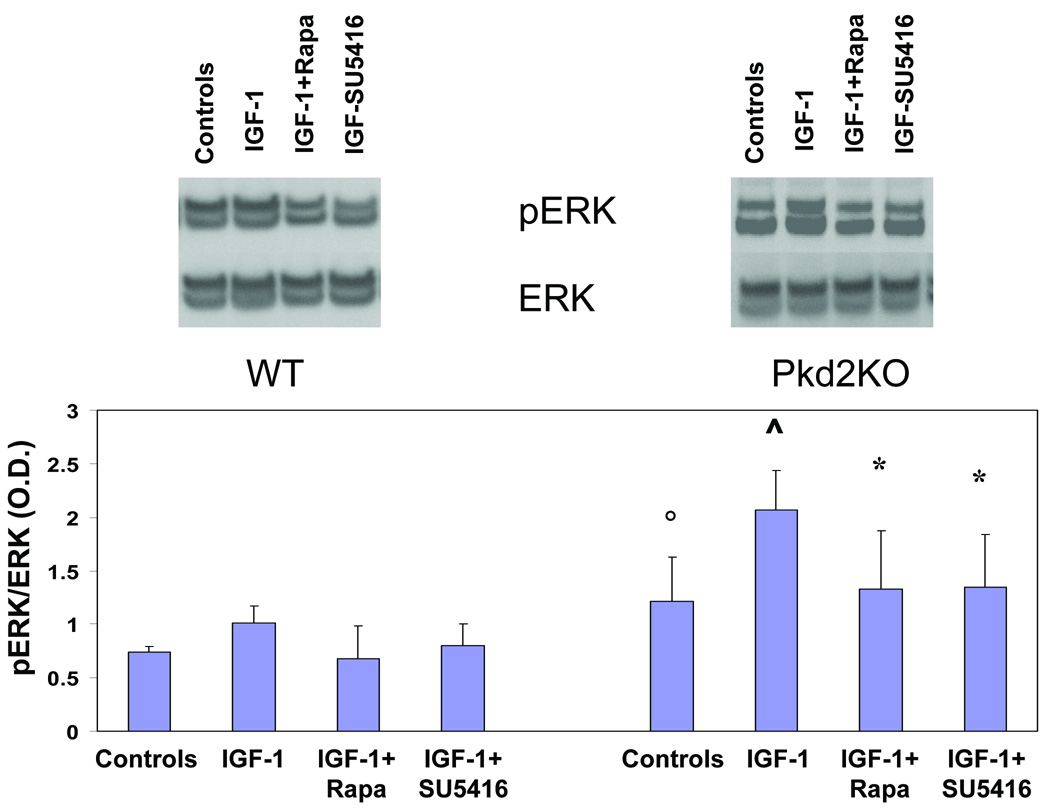
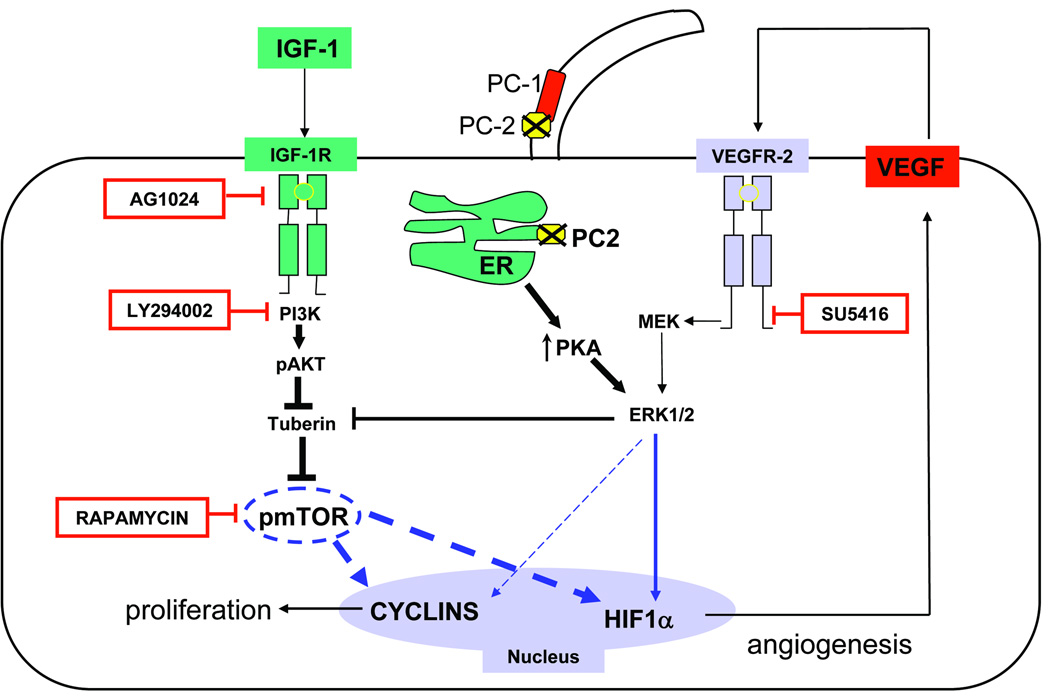
Polycystic liver disease may complicate autosomal dominant polycystic kidney disease (ADPKD), a disease caused by mutations in polycystins, which are proteins that regulate signaling, morphogenesis, and differentiation in epithelial cells. The cystic biliary epithelium [liver cystic epithelium (LCE)] secretes vascular endothelial growth factor (VEGF), which promotes liver cyst growth via autocrine and paracrine mechanisms. The expression of insulin-like growth factor 1 (IGF1), insulin-like growth factor 1 receptor (IGF1R), and phosphorylated mammalian target of rapamycin (p-mTOR) and the protein kinase A (PKA)-dependent phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) are also up-regulated in LCE. We have hypothesized that mammalian target of rapamycin (mTOR) represents a common pathway for the regulation of hypoxia-inducible factor 1 alpha (HIF1alpha)-dependent VEGF secretion by IGF1 and ERK1/2. Conditional polycystin-2-knockout (Pkd2KO) mice were used for in vivo studies and to isolate cystic cholangiocytes [liver cystic epithelial cells (LCECs)]. The expression of p-mTOR, VEGF, cleaved caspase 3 (CC3), proliferating cell nuclear antigen (PCNA), IGF1, IGF1R, phosphorylated extracellular signal-regulated kinase, p-P70S6K, HIF1alpha, and VEGF in LCE, LCECs, and wild-type cholangiocytes was studied with immunohistochemistry, western blotting, or enzyme-linked immunosorbent assays. The cystic area was measured by computer-assisted morphometry of pancytokeratin-stained sections. Cell proliferation in vitro was studied with 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium and bromodeoxyuridine assays. The treatment of Pkd2KO mice with the mTOR inhibitor rapamycin significantly reduced the liver cyst area, liver/body weight ratio, pericystic microvascular density, and PCNA expression while increasing expression of CC3. Rapamycin inhibited IGF1-stimulated HIF1alpha accumulation and VEGF secretion in LCECs. IGF1-stimulated LCEC proliferation was inhibited by rapamycin and SU5416 (a vascular endothelial growth factor receptor 2 inhibitor). Phosphorylation of the mTOR-dependent kinase P70S6K was significantly reduced by PKA inhibitor 14-22 amide and by the mitogen signal-regulated kinase inhibitor U1026.
Conclusion: These data demonstrate that PKA-dependent up-regulation of mTOR has a central role in the proliferative, antiapoptotic, and pro-angiogenic effects of IGF1 and VEGF in polycystin-2-defective mice. This study also highlights a mechanistic link between PKA, ERK, mTOR, and HIF1alpha-mediated VEGF secretion and provides a proof of concept for the potential use of mTOR inhibitors in ADPKD and conditions with aberrant cholangiocyte proliferation.
Conflict of interest statement
All Authors state there are no conflicts of interest to disclose
Figures
References
-
- Bae KT, Zhu F, Chapman AB, Torres VE, Grantham JJ, Guay-Woodford LM, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–69. Epub 2005 Oct 26. - PubMed
-
- Torres VE, Harris PC. Mechanisms of Disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. quiz 55. - PubMed
-
- Li Y, Wright JM, Qian F, Germino GG, Guggino WB. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–41306. Epub 2005 Oct 13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous