

TagRecon: high-throughput mutation identification through sequence tagging
- PMID: 20131910
- PMCID: PMC2859315
- DOI: 10.1021/pr900850m
TagRecon: high-throughput mutation identification through sequence tagging
Abstract
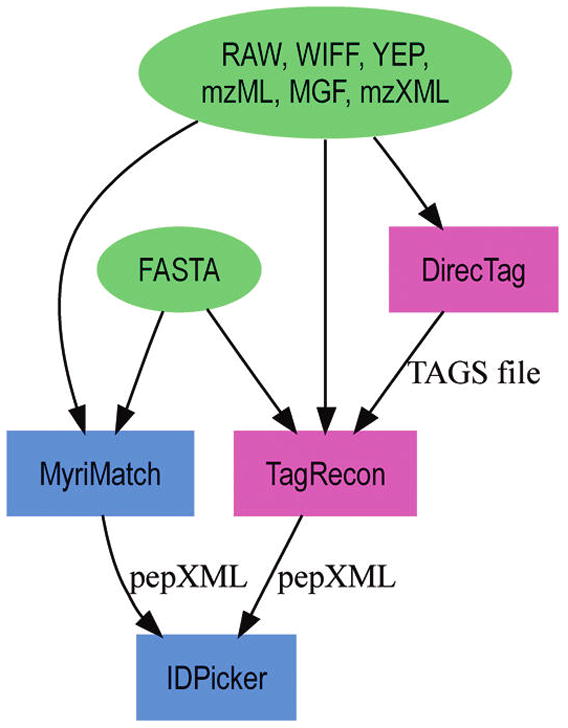
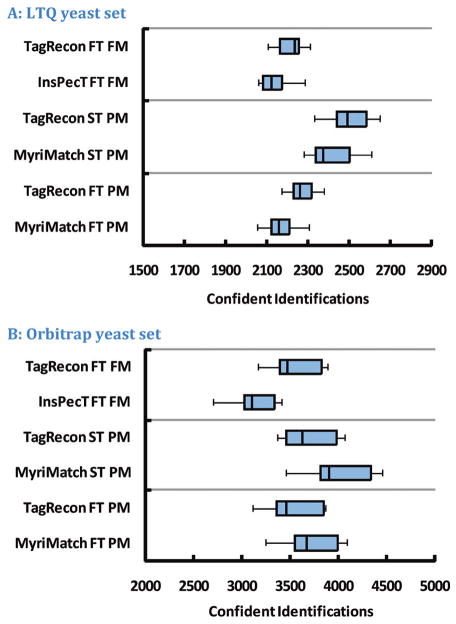
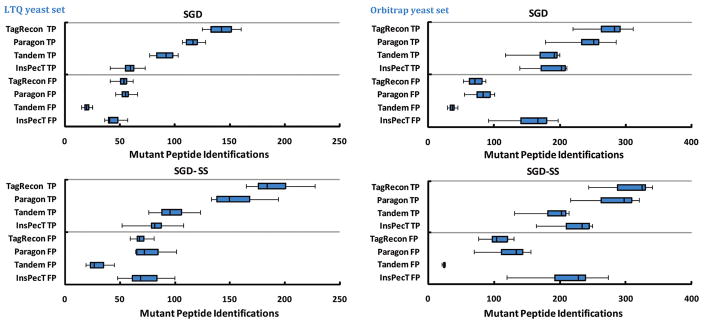
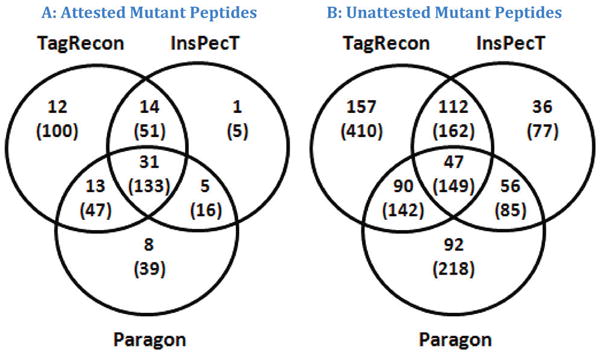
Shotgun proteomics produces collections of tandem mass spectra that contain all the data needed to identify mutated peptides from clinical samples. Identifying these sequence variations, however, has not been feasible with conventional database search strategies, which require exact matches between observed and expected sequences. Searching for mutations as mass shifts on specified residues through database search can incur significant performance penalties and generate substantial false positive rates. Here we describe TagRecon, an algorithm that leverages inferred sequence tags to identify unanticipated mutations in clinical proteomic data sets. TagRecon identifies unmodified peptides as sensitively as the related MyriMatch database search engine. In both LTQ and Orbitrap data sets, TagRecon outperformed state of the art software in recognizing sequence mismatches from data sets with known variants. We developed guidelines for filtering putative mutations from clinical samples, and we applied them in an analysis of cancer cell lines and an examination of colon tissue. Mutations were found in up to 6% of identified peptides, and only a small fraction corresponded to dbSNP entries. The RKO cell line, which is DNA mismatch repair deficient, yielded more mutant peptides than the mismatch repair proficient SW480 line. Analysis of colon cancer tumor and adjacent tissue revealed hydroxyproline modifications associated with extracellular matrix degradation. These results demonstrate the value of using sequence tagging algorithms to fully interrogate clinical proteomic data sets.
Figures

References
-
- Bern M, Goldberg D, McDonald WH, Yates JR. Bioinformatics. 2004;20(Suppl 1):49–54. - PubMed
-
- Nesvizhskii AI, Roos FF, Grossmann J, Vogelzang M, Eddes JS, Gruissem W, Baginsky S, Aebersold R. Mol Cell Proteomics. 2006;5:652–670. - PubMed
-
- Zhao G, Yang F, Yuan Y, Gao X, Zhang J. Yichuan. 2005;27:123–129. - PubMed
-
- Nedelkov D. Expert Rev Proteomics. 2005;2:315–324. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
