

Mechanisms in the loss of capillaries in systemic sclerosis: angiogenesis versus vasculogenesis
- PMID: 20132409
- PMCID: PMC3828842
- DOI: 10.1111/j.1582-4934.2010.01027.x
Mechanisms in the loss of capillaries in systemic sclerosis: angiogenesis versus vasculogenesis
Abstract
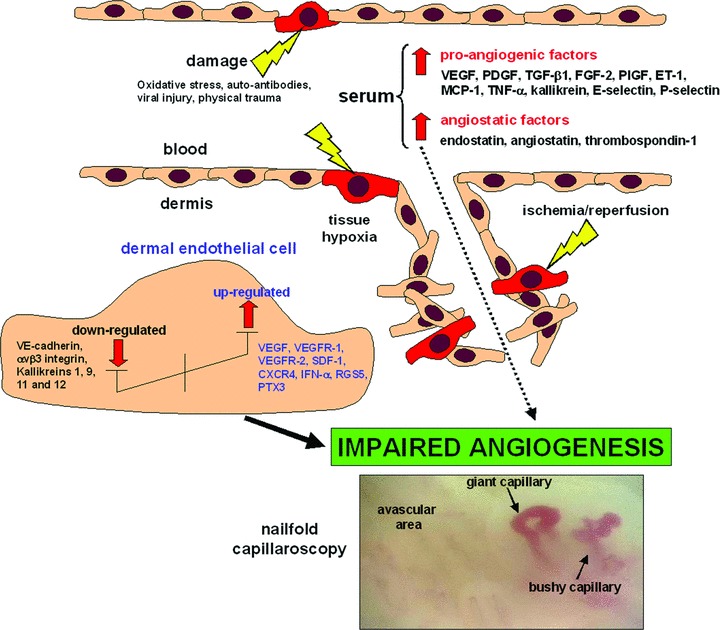
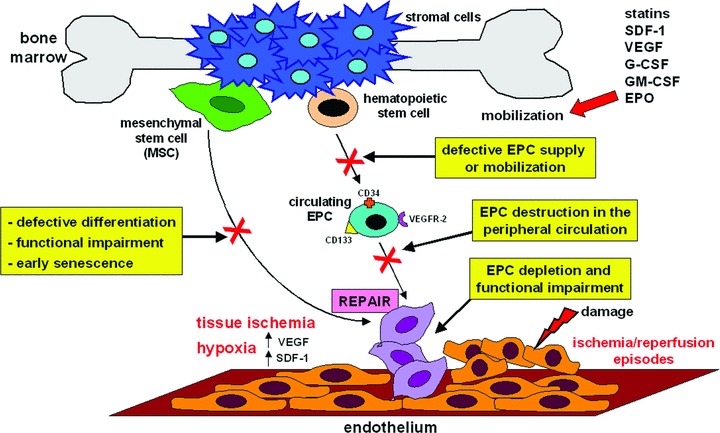
Systemic sclerosis (SSc, scleroderma) is a chronic, multisystem connective tissue disorder affecting the skin and various internal organs. Although the disease is characterized by a triad of widespread microangiopathy, fibrosis and autoimmunity, increasing evidence indicates that vascular damage is a primary event in the pathogenesis of SSc. The progressive vascular injury includes persistent endothelial cell activation/damage and apoptosis, intimal thickening, delamination, vessel narrowing and obliteration. These profound vascular changes lead to vascular tone dysfunction and reduced capillary blood flow, with consequent tissue ischemia and severe clinical manifestations, such as digital ulceration or amputation, pulmonary arterial hypertension and scleroderma renal crisis. The resulting tissue hypoxia induces complex cellular and molecular mechanisms in the attempt to recover endothelial cell function and tissue perfusion. Nevertheless, in SSc patients there is no evidence of significant angiogenesis and the disease evolves towards chronic tissue ischemia, with progressive and irreversible structural changes in multiple vascular beds culminating in the loss of capillaries. A severe imbalance between pro-angiogenic and angiostatic factors may also lead to impaired angiogenic response during SSc. Besides insufficient angiogenesis, defective vasculogenesis with altered numbers and functional defects of bone marrow-derived endothelial progenitor cells may contribute to the vascular pathogenesis of SSc. The purpose of this article is to review the contribution of recent studies to the understanding of the complex mechanisms of impaired vascular repair in SSc. Indeed, understanding the pathophysiology of SSc-associated vascular disease may be the key in dissecting the disease pathogenesis and developing novel therapies. Either angiogenic or vasculogenic mechanisms may potentially become in the future the target of therapeutic strategies to promote capillary regeneration in SSc.
Figures

Similar articles
-
Cellular players in angiogenesis during the course of systemic sclerosis.Autoimmun Rev. 2011 Aug;10(10):641-6. doi: 10.1016/j.autrev.2011.04.016. Epub 2011 Apr 22. Autoimmun Rev. 2011. PMID: 21549220 Review.
-
Impaired angiogenesis in systemic sclerosis: the emerging role of the antiangiogenic VEGF(165)b splice variant.Trends Cardiovasc Med. 2011 Oct;21(7):204-10. doi: 10.1016/j.tcm.2012.05.011. Trends Cardiovasc Med. 2011. PMID: 22867700 Review.
-
The Role of Endothelial Progenitors in the Repair of Vascular Damage in Systemic Sclerosis.Front Immunol. 2018 Jun 18;9:1383. doi: 10.3389/fimmu.2018.01383. eCollection 2018. Front Immunol. 2018. PMID: 29967618 Free PMC article. Review.
-
Systemic Sclerosis Sera Impair Angiogenic Performance of Dermal Microvascular Endothelial Cells: Therapeutic Implications of Cyclophosphamide.PLoS One. 2015 Jun 15;10(6):e0130166. doi: 10.1371/journal.pone.0130166. eCollection 2015. PLoS One. 2015. PMID: 26076019 Free PMC article.
-
Circulating endothelial progenitor cells in systemic sclerosis: relation to impaired angiogenesis and cardiovascular manifestations.Clin Exp Rheumatol. 2008 May-Jun;26(3):421-9. Clin Exp Rheumatol. 2008. PMID: 18578963
Cited by
-
Decreased expression of the endothelial cell-derived factor EGFL7 in systemic sclerosis: potential contribution to impaired angiogenesis and vasculogenesis.Arthritis Res Ther. 2013 Oct 25;15(5):R165. doi: 10.1186/ar4349. Arthritis Res Ther. 2013. PMID: 24286167 Free PMC article.
-
Assessing nailfold microvascular structure with ultra-wideband raster-scan optoacoustic mesoscopy.Photoacoustics. 2018 Feb 21;10:31-37. doi: 10.1016/j.pacs.2018.02.002. eCollection 2018 Jun. Photoacoustics. 2018. PMID: 29988835 Free PMC article.
-
The "myth" of loss of angiogenesis in systemic sclerosis: a pivotal early pathogenetic process or just a late unavoidable event?Arthritis Res Ther. 2017 Jul 6;19(1):162. doi: 10.1186/s13075-017-1370-5. Arthritis Res Ther. 2017. PMID: 28683836 Free PMC article.
-
Molecular Changes Implicate Angiogenesis and Arterial Remodeling in Systemic Sclerosis-Associated and Idiopathic Pulmonary Hypertension.Arterioscler Thromb Vasc Biol. 2024 Aug;44(8):e210-e225. doi: 10.1161/ATVBAHA.123.320005. Epub 2024 Jun 6. Arterioscler Thromb Vasc Biol. 2024. PMID: 38841857 Free PMC article.
-
Neutrophil Extracellular Traps Generation Relates with Early Stage and Vascular Complications in Systemic Sclerosis.J Clin Med. 2020 Jul 7;9(7):2136. doi: 10.3390/jcm9072136. J Clin Med. 2020. PMID: 32645862 Free PMC article.
References
-
- Jimenez SA, Derk CT. Following the molecular pathways towards an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. - PubMed
-
- Guiducci S, Giacomelli R, Matucci-Cerinic M. Vascular complications of scleroderma. Autoimmun Rev. 2007;6:520–3. - PubMed
-
- Kahaleh MB. Vascular involvement in systemic sclerosis. Clin Exp Rheumatol. 2004;22:S19–23. - PubMed
-
- LeRoy EC. Systemic sclerosis: a vascular perspective. Rheum Dis Clin North Am. 1996;22:675–94. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
