

Digital transcriptome profiling from attomole-level RNA samples
- PMID: 20133332
- PMCID: PMC2847755
- DOI: 10.1101/gr.102129.109
Digital transcriptome profiling from attomole-level RNA samples
Abstract
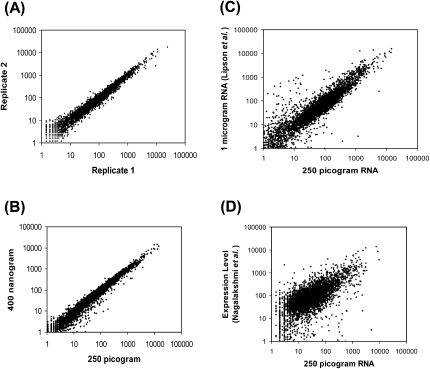
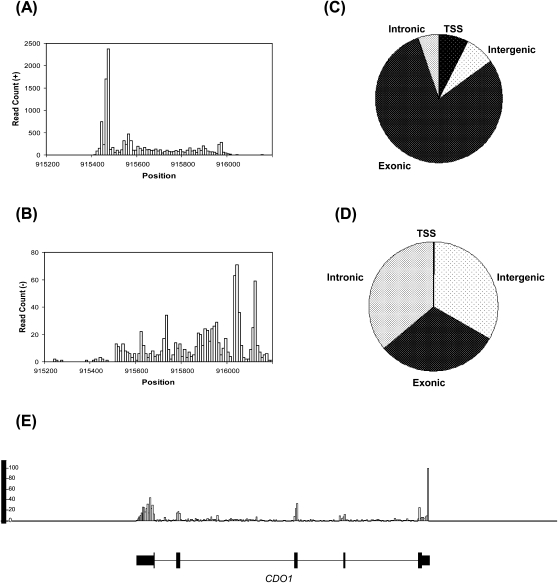
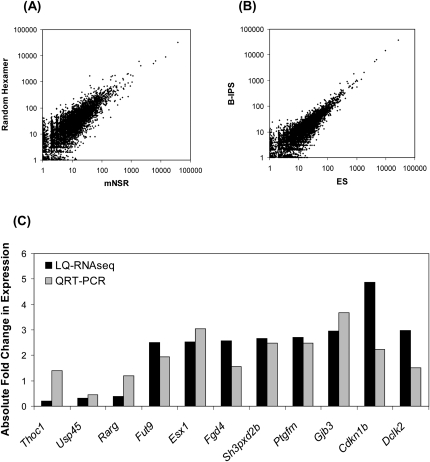
Accurate profiling of minute quantities of RNA in a global manner can enable key advances in many scientific and clinical disciplines. Here, we present low-quantity RNA sequencing (LQ-RNAseq), a high-throughput sequencing-based technique allowing whole transcriptome surveys from subnanogram RNA quantities in an amplification/ligation-free manner. LQ-RNAseq involves first-strand cDNA synthesis from RNA templates, followed by 3' polyA tailing of the single-stranded cDNA products and direct single molecule sequencing. We applied LQ-RNAseq to profile S. cerevisiae polyA+ transcripts, demonstrate the reproducibility of the approach across different sample preparations and independent instrument runs, and establish the absolute quantitative power of this method through comparisons with other reported transcript profiling techniques and through utilization of RNA spike-in experiments. We demonstrate the practical application of this approach to define the transcriptional landscape of mouse embryonic and induced pluripotent stem cells, observing transcriptional differences, including over 100 genes exhibiting differential expression between these otherwise very similar stem cell populations. This amplification-independent technology, which utilizes small quantities of nucleic acid and provides quantitative measurements of cellular transcripts, enables global gene expression measurements from minute amounts of materials and offers broad utility in both basic research and translational biology for characterization of rare cells.
Figures

References
-
- Armour CD, Castle JC, Chen R, Babak T, Loerch P, Jackson S, Shah JK, Dey J, Rohl CA, Johnson JM, et al. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods. 2009;6:647–649. - PubMed
-
- Che S, Ginsberg SD. Amplification of RNA transcripts using terminal continuation. Lab Invest. 2004;84:131–137. - PubMed
-
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods. 2008;5:613–619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases