

Dynamic changes in the human methylome during differentiation
- PMID: 20133333
- PMCID: PMC2840979
- DOI: 10.1101/gr.101907.109
Dynamic changes in the human methylome during differentiation
Abstract
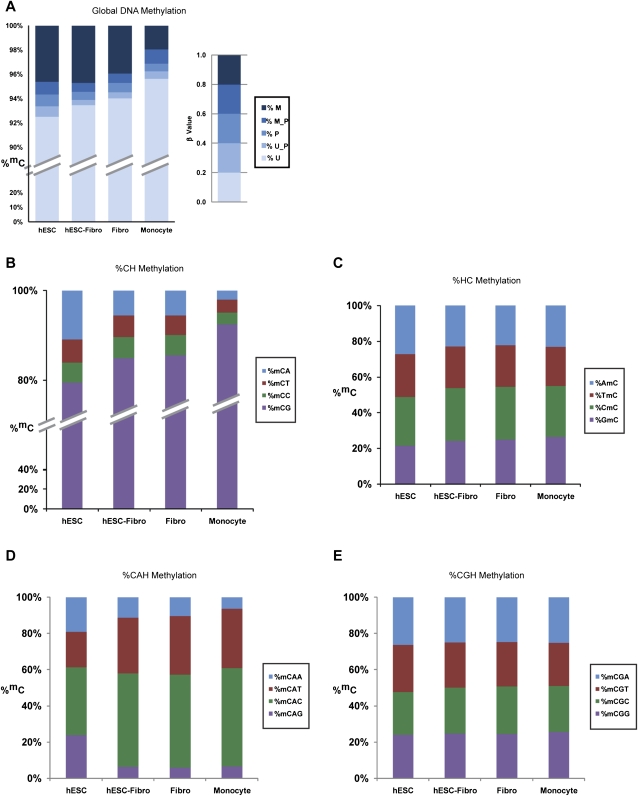
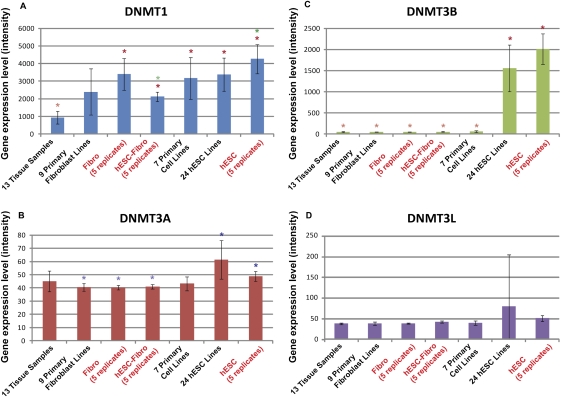
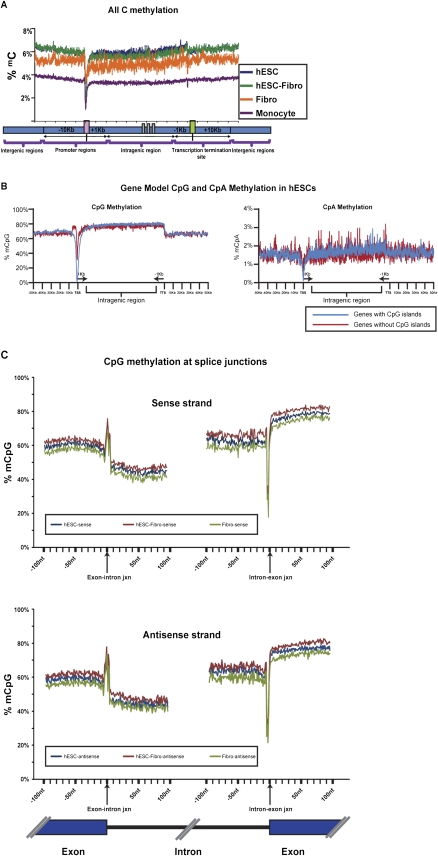
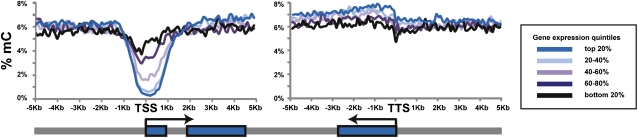
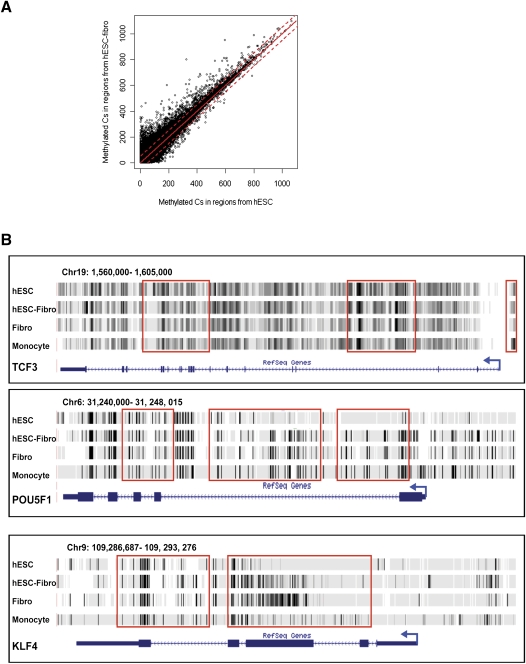
DNA methylation is a critical epigenetic regulator in mammalian development. Here, we present a whole-genome comparative view of DNA methylation using bisulfite sequencing of three cultured cell types representing progressive stages of differentiation: human embryonic stem cells (hESCs), a fibroblastic differentiated derivative of the hESCs, and neonatal fibroblasts. As a reference, we compared our maps with a methylome map of a fully differentiated adult cell type, mature peripheral blood mononuclear cells (monocytes). We observed many notable common and cell-type-specific features among all cell types. Promoter hypomethylation (both CG and CA) and higher levels of gene body methylation were positively correlated with transcription in all cell types. Exons were more highly methylated than introns, and sharp transitions of methylation occurred at exon-intron boundaries, suggesting a role for differential methylation in transcript splicing. Developmental stage was reflected in both the level of global methylation and extent of non-CpG methylation, with hESC highest, fibroblasts intermediate, and monocytes lowest. Differentiation-associated differential methylation profiles were observed for developmentally regulated genes, including the HOX clusters, other homeobox transcription factors, and pluripotence-associated genes such as POU5F1, TCF3, and KLF4. Our results highlight the value of high-resolution methylation maps, in conjunction with other systems-level analyses, for investigation of previously undetectable developmental regulatory mechanisms.
Figures

References
-
- Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol. 2006;13:22–29. - PubMed
-
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. - PubMed
-
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials