

Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling
- PMID: 20133448
- PMCID: PMC2840807
- DOI: 10.1210/me.2009-0357
Deletion of IGF-I receptor (IGF-IR) in primary osteoblasts reduces GH-induced STAT5 signaling
Abstract
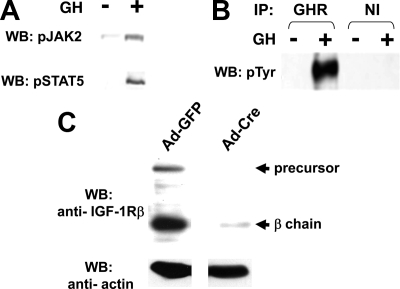
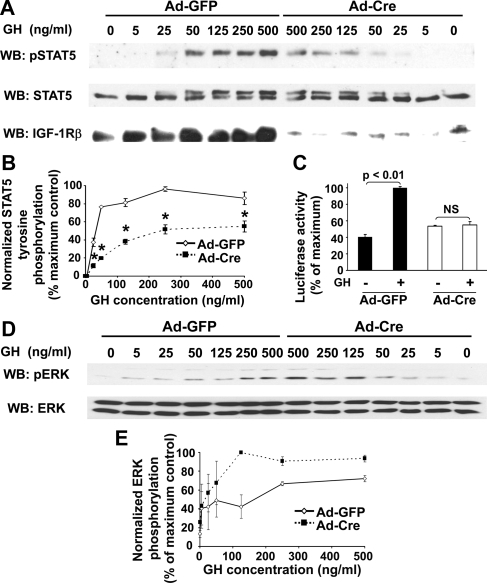
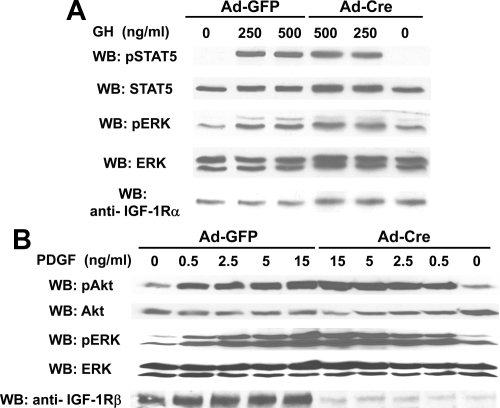
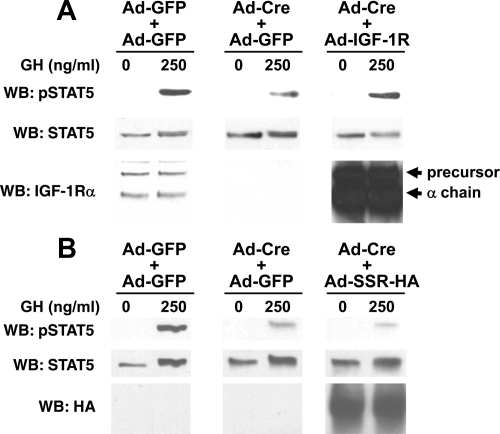
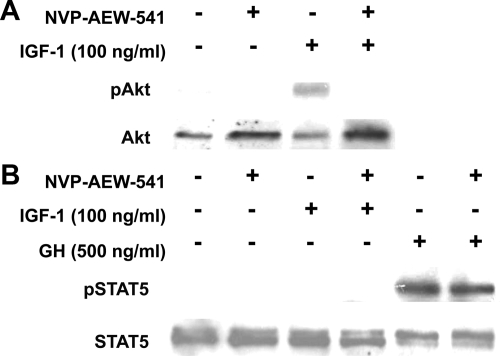
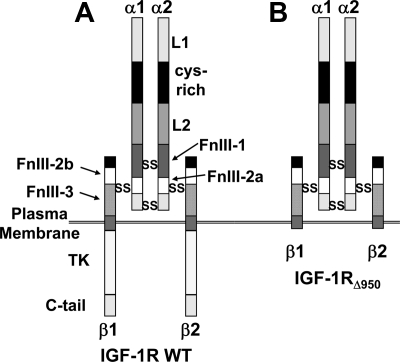
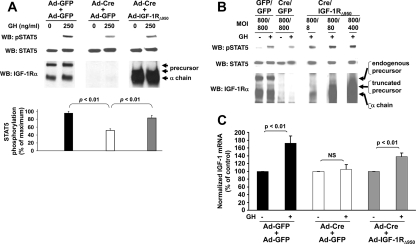
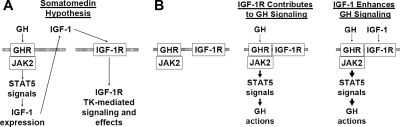
GH promotes longitudinal growth and regulates multiple cellular functions in humans and animals. GH signals by binding to GH receptor (GHR) to activate the tyrosine kinase, Janus kinase 2 (JAK2), and downstream pathways including signal transducer and activator of transcription 5 (STAT5), thereby regulating expression of genes including IGF-I. GH exerts effects both directly and via IGF-I, which signals by activating the IGF-I receptor (IGF-IR). IGF-IR is a cell surface receptor that contains intrinsic tyrosine kinase activity within its intracellular domain. In this study, we examined the potential role of IGF-IR in facilitating GH-induced signal transduction, using mouse primary calvarial osteoblasts with Lox-P sites flanking both IGF-IR alleles. These cells respond to both GH and IGF-I and in vitro infection with an adenovirus that drives expression of Cre recombinase (Ad-Cre) dramatically reduces IGF-IR abundance without affecting the abundance of GHR, JAK2, STAT5, or ERK. Notably, infection with Ad-Cre, but not a control adenovirus, markedly inhibited acute GH-induced STAT5 activity (more than doubling the ED(50) and reducing the maximum activity by nearly 50%), while sparing GH-induced ERK activity, and markedly inhibited GH-induced transactivation of a STAT5-dependent luciferase reporter. The effect of Ad-Cre on GH signaling was specific, as platelet-derived growth factor-induced signaling was unaffected by Ad-Cre-mediated reduction of IGF-IR. Ad-Cre-mediated inhibition of GH signaling was reversed by adenoviral reexpression of IGF-IR, but not by infection with an adenovirus that drives expression of a hemagglutination-tagged somatostatin receptor, which drives expression of the unrelated somatostatin receptor, and Ad-Cre infection of nonfloxed osteoblasts did not affect GH signaling. Notably, infection with an adenovirus encoding a C-terminally truncated IGF-IR that lacks the tyrosine kinase domain partially rescued both acute GH-induced STAT5 activity and GH-induced IGF-I gene expression in cells in which endogenous IGF-IR was reduced. These data, in concert with our earlier findings that GH induces a GHR-JAK2-IGF-IR complex, suggest a novel function for IGF-IR. In addition to its role as a key IGF-I signal transducer, this receptor may directly facilitate acute GH signaling. The implications of these findings are discussed.
Figures
Similar articles
-
IGF-1R modulation of acute GH-induced STAT5 signaling: role of protein tyrosine phosphatase activity.Mol Endocrinol. 2013 Nov;27(11):1969-79. doi: 10.1210/me.2013-1178. Epub 2013 Sep 12. Mol Endocrinol. 2013. PMID: 24030252 Free PMC article.
-
Functional collaboration of insulin-like growth factor-1 receptor (IGF-1R), but not insulin receptor (IR), with acute GH signaling in mouse calvarial cells.Endocrinology. 2014 Mar;155(3):1000-9. doi: 10.1210/en.2013-1732. Epub 2013 Jan 1. Endocrinology. 2014. PMID: 24302626 Free PMC article.
-
Human GH receptor-IGF-1 receptor interaction: implications for GH signaling.Mol Endocrinol. 2014 Nov;28(11):1841-54. doi: 10.1210/me.2014-1174. Epub 2014 Sep 11. Mol Endocrinol. 2014. PMID: 25211187 Free PMC article.
-
Human growth disorders associated with impaired GH action: Defects in STAT5B and JAK2.Mol Cell Endocrinol. 2021 Jan 1;519:111063. doi: 10.1016/j.mce.2020.111063. Epub 2020 Oct 27. Mol Cell Endocrinol. 2021. PMID: 33122102 Free PMC article. Review.
-
Minireview: mechanisms of growth hormone-mediated gene regulation.Mol Endocrinol. 2014 Jul;28(7):1012-25. doi: 10.1210/me.2014-1099. Epub 2014 May 13. Mol Endocrinol. 2014. PMID: 24825400 Free PMC article. Review.
Cited by
-
DMP-1-mediated Ghr gene recombination compromises skeletal development and impairs skeletal response to intermittent PTH.FASEB J. 2016 Feb;30(2):635-52. doi: 10.1096/fj.15-275859. Epub 2015 Oct 19. FASEB J. 2016. PMID: 26481310 Free PMC article.
-
IGF-1R modulation of acute GH-induced STAT5 signaling: role of protein tyrosine phosphatase activity.Mol Endocrinol. 2013 Nov;27(11):1969-79. doi: 10.1210/me.2013-1178. Epub 2013 Sep 12. Mol Endocrinol. 2013. PMID: 24030252 Free PMC article.
-
Functional collaboration of insulin-like growth factor-1 receptor (IGF-1R), but not insulin receptor (IR), with acute GH signaling in mouse calvarial cells.Endocrinology. 2014 Mar;155(3):1000-9. doi: 10.1210/en.2013-1732. Epub 2013 Jan 1. Endocrinology. 2014. PMID: 24302626 Free PMC article.
-
MSM enhances GH signaling via the Jak2/STAT5b pathway in osteoblast-like cells and osteoblast differentiation through the activation of STAT5b in MSCs.PLoS One. 2012;7(10):e47477. doi: 10.1371/journal.pone.0047477. Epub 2012 Oct 11. PLoS One. 2012. PMID: 23071812 Free PMC article.
-
Effects of leptin on pedunculopontine nucleus (PPN) neurons.J Neural Transm (Vienna). 2013 Jul;120(7):1027-38. doi: 10.1007/s00702-012-0957-x. Epub 2012 Dec 21. J Neural Transm (Vienna). 2013. PMID: 23263542 Free PMC article.
References
-
- Isaksson OG, Eden S, Jansson JO 1985 Mode of action of pituitary growth hormone on target cells. Annu Rev Physiol 47:483–499 - PubMed
-
- Kaplan S 1999 Hormonal regulation of growth and metabolic effects of growth hormone. In: Kostyo J, Goodman HM, eds. Handbook of physiology. New York: Oxford University Press; 129–143
-
- Frank SJ, Messina JL 2002 Growth hormone receptor. In: Oppenheim JJ, Feldman M, eds. Cytokine reference on-line. London, UK: Academic Press, Harcourt; 1–21
-
- Carter Su C, Schwartz J, Smit LS 1996 Molecular mechanism of growth hormone action. Annu Rev Physiol 58:187–207 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
