

Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis
- PMID: 20133622
- PMCID: PMC2840338
- DOI: 10.1073/pnas.0910533107
Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis
Abstract
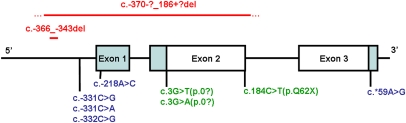
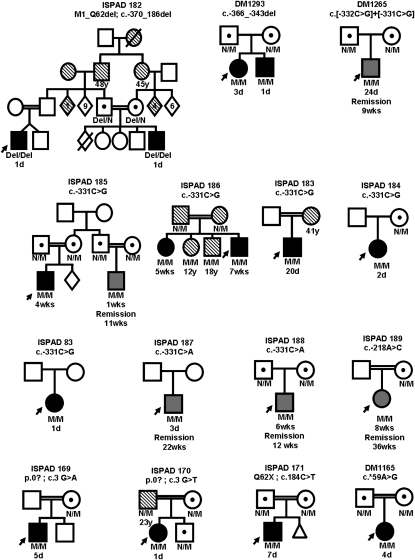
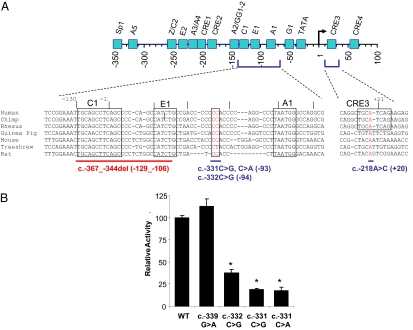
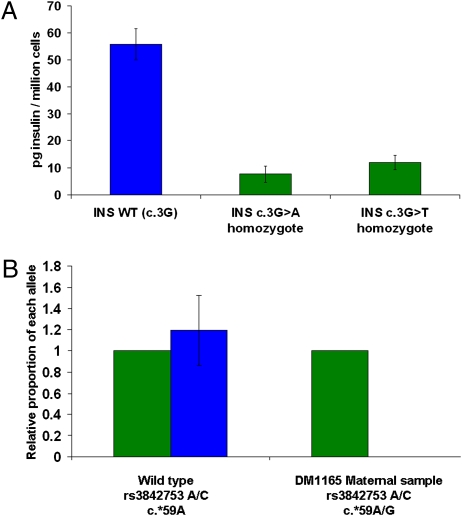
Heterozygous coding mutations in the INS gene that encodes preproinsulin were recently shown to be an important cause of permanent neonatal diabetes. These dominantly acting mutations prevent normal folding of proinsulin, which leads to beta-cell death through endoplasmic reticulum stress and apoptosis. We now report 10 different recessive INS mutations in 15 probands with neonatal diabetes. Functional studies showed that recessive mutations resulted in diabetes because of decreased insulin biosynthesis through distinct mechanisms, including gene deletion, lack of the translation initiation signal, and altered mRNA stability because of the disruption of a polyadenylation signal. A subset of recessive mutations caused abnormal INS transcription, including the deletion of the C1 and E1 cis regulatory elements, or three different single base-pair substitutions in a CC dinucleotide sequence located between E1 and A1 elements. In keeping with an earlier and more severe beta-cell defect, patients with recessive INS mutations had a lower birth weight (-3.2 SD score vs. -2.0 SD score) and were diagnosed earlier (median 1 week vs. 10 weeks) compared to those with dominant INS mutations. Mutations in the insulin gene can therefore result in neonatal diabetes as a result of two contrasting pathogenic mechanisms. Moreover, the recessively inherited mutations provide a genetic demonstration of the essential role of multiple sequence elements that regulate the biosynthesis of insulin in man.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Iafusco D, et al. Early Onset Diabetes Study Group of the Italian Society of Paediatric Endocrinology and Diabetology. Permanent diabetes mellitus in the first year of life. Diabetologia. 2002;45:798–804. - PubMed
-
- Edghill EL, et al. HLA genotyping supports a nonautoimmune etiology in patients diagnosed with diabetes under the age of 6 months. Diabetes. 2006;55:1895–1898. - PubMed
-
- Gloyn AL, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. - PubMed
-
- Proks P, et al. A heterozygous activating mutation in the sulphonylurea receptor SUR1 (ABCC8) causes neonatal diabetes. Hum Mol Genet. 2006;15:1793–1800. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
