

STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress
- PMID: 20133729
- PMCID: PMC2840303
- DOI: 10.1073/pnas.0909086107
STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress
Abstract
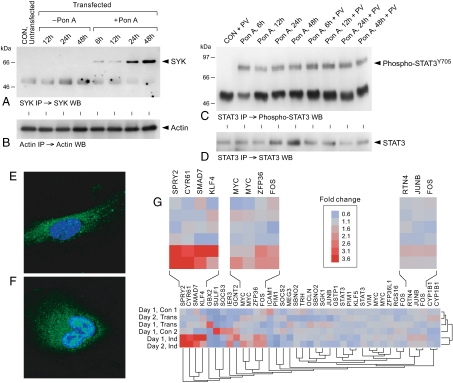
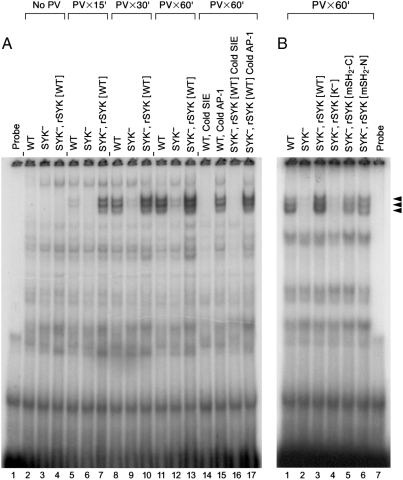
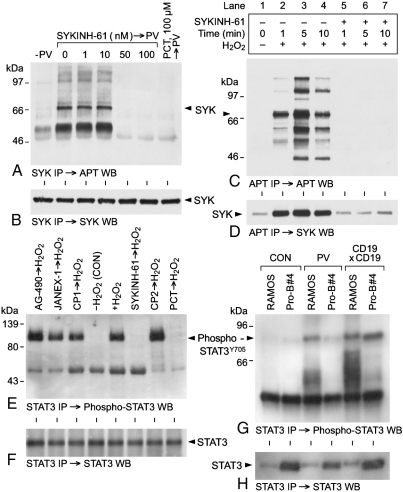
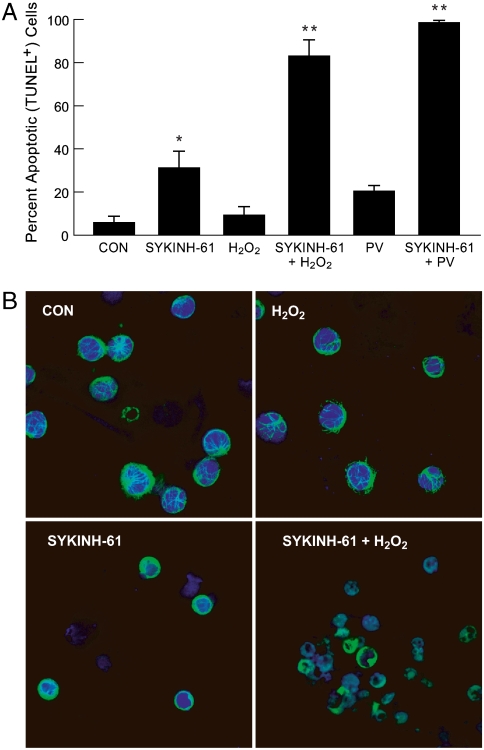
We provide unprecedented genetic and biochemical evidence that the antiapoptotic transcription factor STAT3 serves as a substrate for SYK tyrosine kinase both in vitro and in vivo. Induction of SYK in an ecdysone-inducible mammalian expression system results in STAT3 activation, as documented by tyrosine phosphorylation and nuclear translocation of STAT3, as well as amplified expression of several STAT3 target genes. STAT3 activation after oxidative stress (OS) is strongly diminished in DT40 chicken B-lineage lymphoma cells rendered SYK-deficient by targeted disruption of the syk gene. Introduction of a wild-type, C-terminal or N-terminal SH2 domain-mutated, but not a kinase domain-mutated, syk gene into SYK-deficient DT40 cells restores OS-induced enhancement of STAT-3 activity. Thus, SYK plays an important and indispensable role in OS-induced STAT3 activation and its catalytic SH1 domain is critical for this previously unknown regulatory function. These results provide evidence for the existence of a novel mode of cytokine-independent cross-talk that operates between SYK and STAT3 pathways and regulates apoptosis during OS. We further provide experimental evidence that SYK is capable of associating with and phosphorylating STAT3 in human B-lineage leukemia/lymphoma cells challenged with OS. In agreement with a prerequisite role of SYK in OS-induced STAT3 activation, OS does not induce tyrosine phosphorylation of STAT3 in SYK-deficient human proB leukemia cells. Notably, inhibition of SYK with a small molecule drug candidate prevents OS-induced activation of STAT3 and overcomes the resistance of human B-lineage leukemia/lymphoma cells to OS-induced apoptosis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Polo-like-kinase 1 (PLK1) as a molecular target to overcome SYK-mediated resistance of B-lineage acute lymphoblastic leukaemia cells to oxidative stress.Br J Haematol. 2010 Mar;148(5):714-25. doi: 10.1111/j.1365-2141.2009.07983.x. Epub 2009 Nov 12. Br J Haematol. 2010. PMID: 19912216
-
Bruton's tyrosine kinase prevents activation of the anti-apoptotic transcription factor STAT3 and promotes apoptosis in neoplastic B-cells and B-cell precursors exposed to oxidative stress.Br J Haematol. 2007 Feb;136(4):574-89. doi: 10.1111/j.1365-2141.2006.06468.x. Br J Haematol. 2007. PMID: 17367410
-
Stimulation of Src family protein-tyrosine kinases as a proximal and mandatory step for SYK kinase-dependent phospholipase Cgamma2 activation in lymphoma B cells exposed to low energy electromagnetic fields.J Biol Chem. 1998 Feb 13;273(7):4035-9. doi: 10.1074/jbc.273.7.4035. J Biol Chem. 1998. PMID: 9461594
-
Spleen tyrosine kinase as a molecular target for treatment of leukemias and lymphomas.Expert Rev Anticancer Ther. 2010 Sep;10(9):1407-18. doi: 10.1586/era.10.112. Expert Rev Anticancer Ther. 2010. PMID: 20836676 Review.
-
Role of protein-tyrosine kinase syk in oxidative stress signaling in B cells.Antioxid Redox Signal. 2002 Jun;4(3):533-41. doi: 10.1089/15230860260196335. Antioxid Redox Signal. 2002. PMID: 12215221 Review.
Cited by
-
An oxidative stress-based mechanism of doxorubicin cytotoxicity suggests new therapeutic strategies in ABC-DLBCL.Blood. 2016 Dec 15;128(24):2797-2807. doi: 10.1182/blood-2016-03-705814. Epub 2016 Oct 13. Blood. 2016. PMID: 27737889 Free PMC article.
-
Nanomedicines in B cell-targeting therapies.Acta Biomater. 2022 Jan 1;137:1-19. doi: 10.1016/j.actbio.2021.10.024. Epub 2021 Oct 21. Acta Biomater. 2022. PMID: 34687954 Free PMC article. Review.
-
Elevated mitochondrial superoxide disrupts normal T cell development, impairing adaptive immune responses to an influenza challenge.Free Radic Biol Med. 2011 Feb 1;50(3):448-58. doi: 10.1016/j.freeradbiomed.2010.11.025. Epub 2010 Dec 2. Free Radic Biol Med. 2011. PMID: 21130157 Free PMC article.
-
Contemporary patient-tailored treatment strategies against high risk and relapsed or refractory multiple myeloma.EBioMedicine. 2019 Jan;39:612-620. doi: 10.1016/j.ebiom.2018.12.004. Epub 2018 Dec 10. EBioMedicine. 2019. PMID: 30545798 Free PMC article. Review.
-
Reelin promotes the adhesion and drug resistance of multiple myeloma cells via integrin β1 signaling and STAT3.Oncotarget. 2016 Mar 1;7(9):9844-58. doi: 10.18632/oncotarget.7151. Oncotarget. 2016. PMID: 26848618 Free PMC article.
References
-
- Trigg ME, Gaynon P, Uckun FM. In: Cancer Medicine. 4th Ed. Holland JF, et al., editors. London: B.C. Decker, Inc.; 1996. pp. 2945–2960.
-
- Gaynon PS, et al. Bone marrow transplantation versus prolonged intensive chemotherapy for children with acute lymphoblastic leukemia and an initial bone marrow relapse within 12 months of the completion of primary therapy: Children’s Oncology Group Study CCG-1941. J Clin Oncol. 2006;24:3150–3156. - PubMed
-
- Bailey LC, Lange BJ, Rheinhold SR, Bunin NJ. Bone marrow relapse in paediatric acute lymphoblastic leukemia. Lancet Oncol. 2008;9(9):873–83. - PubMed
-
- Weston VJ, et al. Apoptotic resistance to ionizing radiation in pediatric B-precursor acute lymphoblastic leukemia frequently involves increased NF kappa B survival pathway signaling. Blood. 2004;104:1465–1473. - PubMed
-
- Marston E, et al. Stratification of pediatric ALL by in vitro cellular responses to DNA double-strand breaks provides insight into the molecular mechanisms underlying clinical response. Blood. 2009;113:117–126. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
