

Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism
- PMID: 20133771
- PMCID: PMC2840277
- DOI: 10.1073/pnas.0910717107
Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism
Abstract
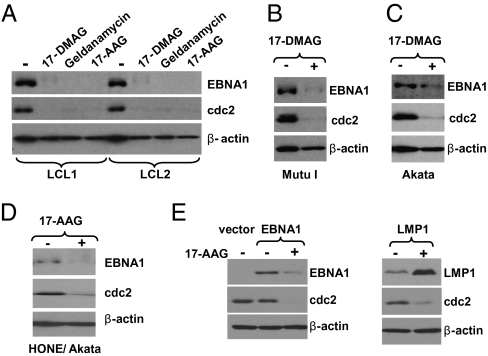
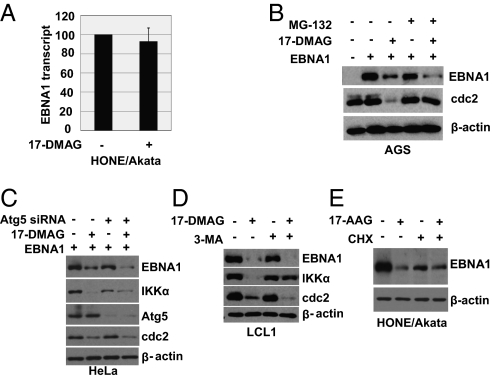
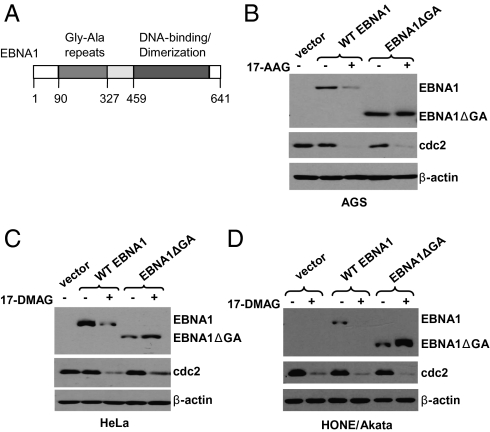
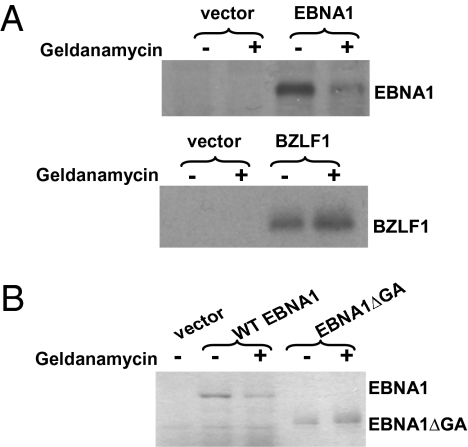
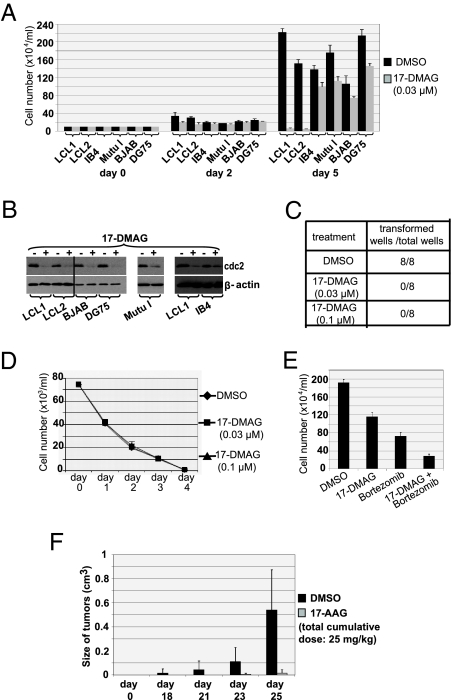
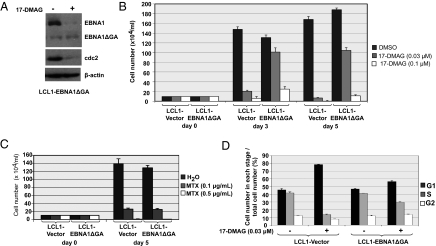
EBV causes infectious mononucleosis and is associated with certain malignancies. EBV nuclear antigen 1 (EBNA1) mediates EBV genome replication, partition, and transcription, and is essential for persistence of the viral genome in host cells. Here we demonstrate that Hsp90 inhibitors decrease EBNA1 expression and translation, and that this effect requires the Gly-Ala repeat domain of EBNA1. Hsp90 inhibitors induce the death of established, EBV-transformed lymphoblastoid cell lines at doses nontoxic to normal cells, and this effect is substantially reversed when lymphoblastoid cell lines are stably infected with a retrovirus expressing a functional EBNA1 mutant lacking the Gly-Ala repeats. Hsp90 inhibitors prevent EBV transformation of primary B cells, and strongly inhibit the growth of EBV-induced lymphoproliferative disease in SCID mice. These results suggest that Hsp90 inhibitors may be particularly effective for treating EBV-induced diseases requiring the continued presence of the viral genome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kieff E, Richinson AB. 2007. Epstein-Barr virus and its replication. Fields’ virology. (Lippincott Williams & Wilkins, Philadelphia) 5th ed. eds Knipe D, etal., pp 2603–2654.
-
- Niller HH, Wolf H, Minarovits J. Regulation and dysregulation of Epstein-Barr virus latency: implications for the development of autoimmune diseases. Autoimmunity. 2008;41:298–328. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
