

Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein
- PMID: 20133840
- PMCID: PMC2808220
- DOI: 10.1073/pnas.0910715107
Structural insights into phosphoinositide 3-kinase activation by the influenza A virus NS1 protein
Abstract
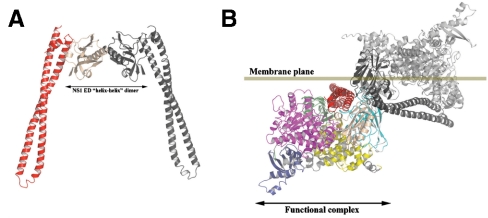
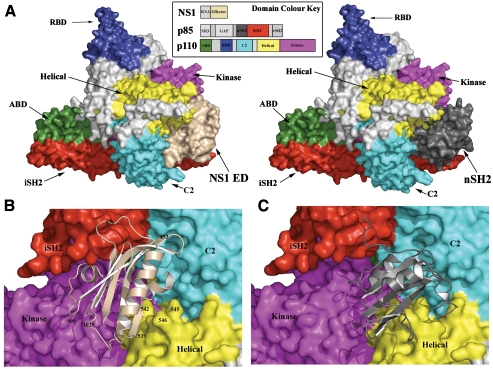
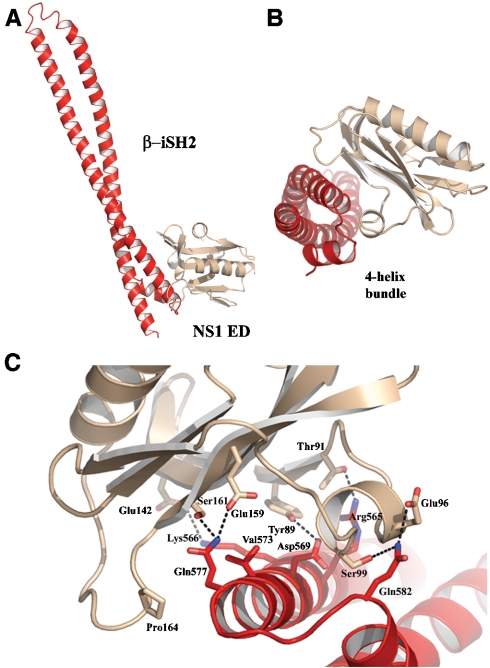
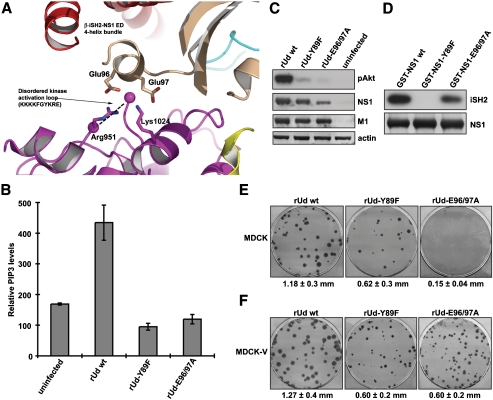
Seasonal epidemics and periodic worldwide pandemics caused by influenza A viruses are of continuous concern. The viral nonstructural (NS1) protein is a multifunctional virulence factor that antagonizes several host innate immune defenses during infection. NS1 also directly stimulates class IA phosphoinositide 3-kinase (PI3K) signaling, an essential cell survival pathway commonly mutated in human cancers. Here, we present a 2.3-A resolution crystal structure of the NS1 effector domain in complex with the inter-SH2 (coiled-coil) domain of p85beta, a regulatory subunit of PI3K. Our data emphasize the remarkable isoform specificity of this interaction, and provide insights into the mechanism by which NS1 activates the PI3K (p85beta:p110) holoenzyme. A model of the NS1:PI3K heterotrimeric complex reveals that NS1 uses the coiled-coil as a structural tether to sterically prevent normal inhibitory contacts between the N-terminal SH2 domain of p85beta and the p110 catalytic subunit. Furthermore, in this model, NS1 makes extensive contacts with the C2/kinase domains of p110, and a small acidic alpha-helix of NS1 sits adjacent to the highly basic activation loop of the enzyme. During infection, a recombinant influenza A virus expressing NS1 with charge-disruption mutations in this acidic alpha-helix is unable to stimulate the production of phosphatidylinositol 3,4,5-trisphosphate or the phosphorylation of Akt. Despite this, the charge-disruption mutations in NS1 do not affect its ability to interact with the p85beta inter-SH2 domain in vitro. Overall, these data suggest that both direct binding of NS1 to p85beta (resulting in repositioning of the N-terminal SH2 domain) and possible NS1:p110 contacts contribute to PI3K activation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Binding of influenza A virus NS1 protein to the inter-SH2 domain of p85 suggests a novel mechanism for phosphoinositide 3-kinase activation.J Biol Chem. 2008 Jan 18;283(3):1372-1380. doi: 10.1074/jbc.M708862200. Epub 2007 Nov 20. J Biol Chem. 2008. PMID: 18029356
-
Mechanism of influenza A virus NS1 protein interaction with the p85beta, but not the p85alpha, subunit of phosphatidylinositol 3-kinase (PI3K) and up-regulation of PI3K activity.J Biol Chem. 2008 Aug 22;283(34):23397-409. doi: 10.1074/jbc.M802737200. Epub 2008 Jun 5. J Biol Chem. 2008. PMID: 18534979
-
Structure-Guided Functional Annotation of the Influenza A Virus NS1 Protein Reveals Dynamic Evolution of the p85β-Binding Site during Circulation in Humans.J Virol. 2017 Oct 13;91(21):e01081-17. doi: 10.1128/JVI.01081-17. Print 2017 Nov 1. J Virol. 2017. PMID: 28814525 Free PMC article.
-
The regulation of class IA PI 3-kinases by inter-subunit interactions.Curr Top Microbiol Immunol. 2010;346:87-114. doi: 10.1007/82_2010_52. Curr Top Microbiol Immunol. 2010. PMID: 20544340 Free PMC article. Review.
-
Capitalizing on tumor genotyping: towards the design of mutation specific inhibitors of phosphoinsitide-3-kinase.Adv Enzyme Regul. 2011;51(1):273-9. doi: 10.1016/j.advenzreg.2010.09.013. Epub 2010 Oct 28. Adv Enzyme Regul. 2011. PMID: 21035489 Free PMC article. Review.
Cited by
-
The Two Sides of the Same Coin-Influenza Virus and Intracellular Signal Transduction.Cold Spring Harb Perspect Med. 2021 Jan 4;11(1):a038513. doi: 10.1101/cshperspect.a038513. Cold Spring Harb Perspect Med. 2021. PMID: 31871235 Free PMC article. Review.
-
Suppressing Nonspecific Binding in Biolayer Interferometry Experiments for Weak Ligand-Analyte Interactions.ACS Omega. 2022 Mar 9;7(11):9206-9211. doi: 10.1021/acsomega.1c05659. eCollection 2022 Mar 22. ACS Omega. 2022. PMID: 35350330 Free PMC article.
-
Survival analysis of infected mice reveals pathogenic variations in the genome of avian H1N1 viruses.Sci Rep. 2014 Dec 12;4:7455. doi: 10.1038/srep07455. Sci Rep. 2014. PMID: 25503687 Free PMC article.
-
New small-molecule drug design strategies for fighting resistant influenza A.Acta Pharm Sin B. 2015 Sep;5(5):419-30. doi: 10.1016/j.apsb.2015.07.006. Epub 2015 Sep 6. Acta Pharm Sin B. 2015. PMID: 26579472 Free PMC article. Review.
-
Molecular Basis of Oncogenic PI3K Proteins.Cancers (Basel). 2024 Dec 30;17(1):77. doi: 10.3390/cancers17010077. Cancers (Basel). 2024. PMID: 39796708 Free PMC article. Review.
References
-
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. - PubMed
-
- Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. - PubMed
-
- Huang CH, et al. The structure of a human p110alpha/p85alpha complex elucidates the effects of oncogenic PI3Kalpha mutations. Science. 2007;318(5857):1744–1748. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
