

Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer
- PMID: 20136112
- PMCID: PMC5330319
- DOI: 10.1021/jp909640g
Free energy profile of the interaction between a monomer or a dimer of protegrin-1 in a specific binding orientation and a model lipid bilayer
Abstract
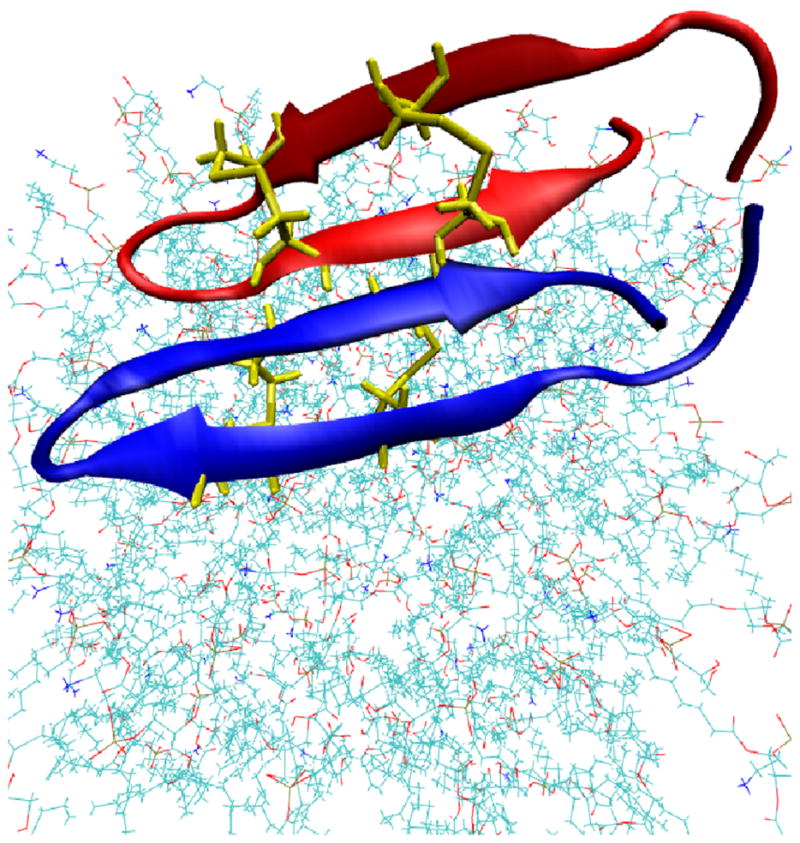
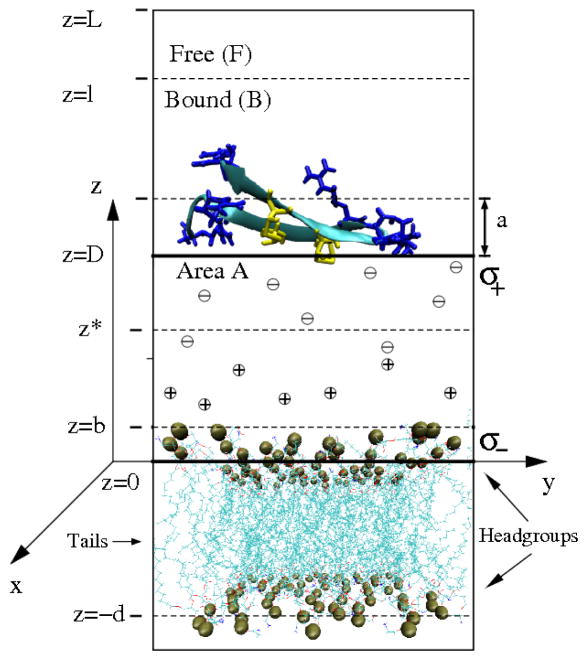
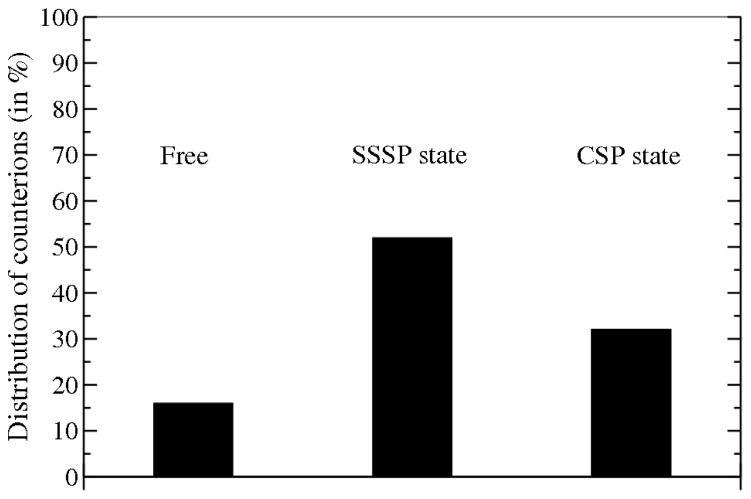
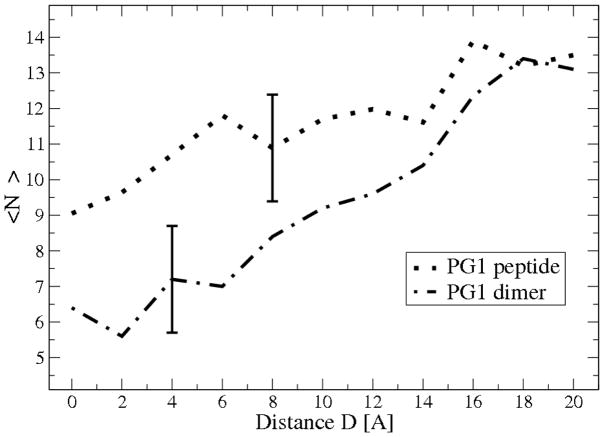
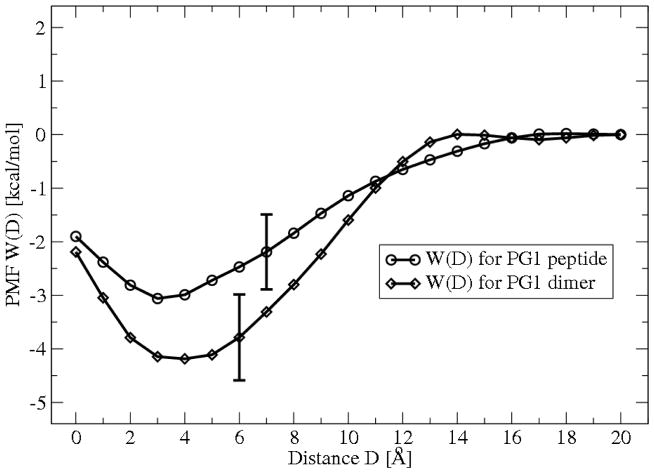
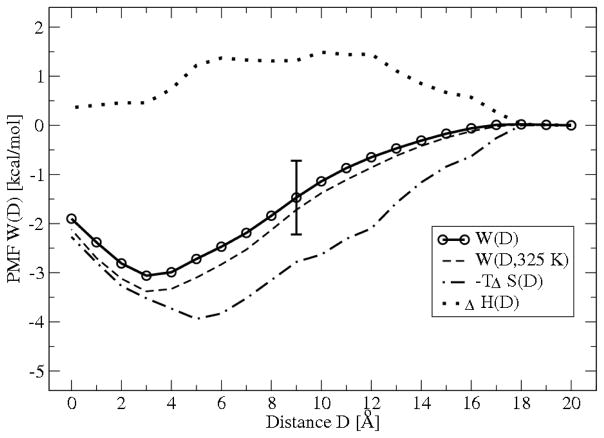
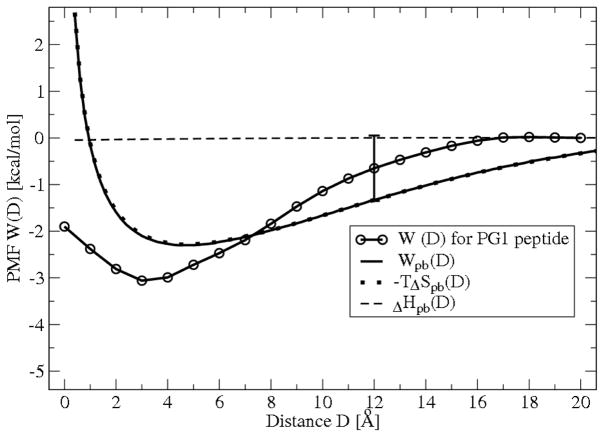
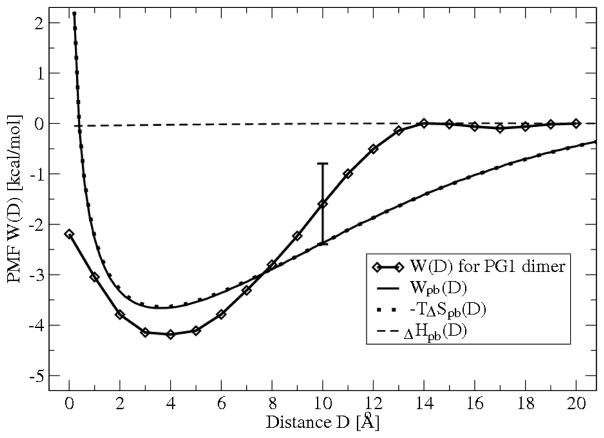
The free energies of adsorption of the monomer or dimer of the cationic beta-hairpin antimicrobial peptide protegrin-1 (PG1) in a specific binding orientation on a lipid bilayer are determined using molecular dynamics (MD) simulations and Poisson-Boltzmann calculations. The bilayer is composed of anionic palmitoyl-oleoyl-phosphatidylglycerol (POPG) and palmitoyl-oleoyl-phosphatidylethanolamine (POPE) with ratio 1:3 (POPG/POPE). PG1 is believed to kill bacteria by binding on their membranes. There, it forms pores that lyse the bacteria. Herein we focus on the thermodynamics of binding. In particular, we explore the role of counterion release from the lipid bilayer upon adsorption of either the monomeric or the dimeric form of PG1. Twenty-two 4-ns-long MD trajectories of equilibrated systems are generated to determine the free energy profiles for the monomer and dimer as a function of the distance between the peptide(s) and the membrane surface. The MD simulations are conducted at 11 different separations from the membrane for each of the two systems, one with PG1, the second with a PG1 dimer of only a specific orientation of the monomer and dimer without taking into account the change of entropy for the peptide. To calculate the potential of mean force for each peptide/membrane system, a variant of constrained MD and thermodynamic integration is used. We observed that PG1 dimer binds more favorably to the POPG/POPE membrane. A simple method for relating the free energy profile to the PG1-membrane binding constant is employed to predict a free energy of adsorption of -2.4 +/- 0.8 kcal/mol. A corresponding PG1-dimer-membrane binding constant is calculated as -3.5 +/- 1.1 kcal/mol. Free energy profiles from MD simulation were extensively analyzed and compared with results of Poisson-Boltzmann theory. We find the peptide-membrane attraction to be dominated by the entropy increase due to the release of counterions in a POPG/POPE lipid bilayer.
Figures
Similar articles
-
Dimerization of protegrin-1 in different environments.Int J Mol Sci. 2010 Sep 9;11(9):3177-94. doi: 10.3390/ijms11093177. Int J Mol Sci. 2010. PMID: 20957087 Free PMC article.
-
Binding free energy and counterion release for adsorption of the antimicrobial peptide lactoferricin B on a POPG membrane.Phys Rev E Stat Nonlin Soft Matter Phys. 2009 Sep;80(3 Pt 1):031911. doi: 10.1103/PhysRevE.80.031911. Epub 2009 Sep 22. Phys Rev E Stat Nonlin Soft Matter Phys. 2009. PMID: 19905150
-
Thermodynamic analysis of protegrin-1 insertion and permeation through a lipid bilayer.J Phys Chem B. 2011 Dec 15;115(49):14704-12. doi: 10.1021/jp205153y. Epub 2011 Nov 18. J Phys Chem B. 2011. PMID: 22044268 Free PMC article.
-
Peptide-lipid interactions of the beta-hairpin antimicrobial peptide tachyplesin and its linear derivatives from solid-state NMR.Biochim Biophys Acta. 2006 Sep;1758(9):1285-91. doi: 10.1016/j.bbamem.2006.03.016. Epub 2006 Apr 5. Biochim Biophys Acta. 2006. PMID: 16678119 Review.
-
An Antimicrobial Peptide-Mimetic Methacrylate Random Copolymer Induces Domain Formation in a Model Bacterial Membrane.J Membr Biol. 2022 Oct;255(4-5):513-521. doi: 10.1007/s00232-022-00220-6. Epub 2022 Feb 18. J Membr Biol. 2022. PMID: 35182193 Review.
Cited by
-
Multiscale models of the antimicrobial peptide protegrin-1 on gram-negative bacteria membranes.Int J Mol Sci. 2012;13(9):11000-11011. doi: 10.3390/ijms130911000. Epub 2012 Sep 5. Int J Mol Sci. 2012. PMID: 23109834 Free PMC article. Review.
-
Dimerization of protegrin-1 in different environments.Int J Mol Sci. 2010 Sep 9;11(9):3177-94. doi: 10.3390/ijms11093177. Int J Mol Sci. 2010. PMID: 20957087 Free PMC article.
-
Multiscale Models of Antibiotic Probiotics.Curr Opin Chem Eng. 2014 Nov 1;6:18-24. doi: 10.1016/j.coche.2014.08.002. Curr Opin Chem Eng. 2014. PMID: 25313349 Free PMC article.
-
Computational studies of peptide-induced membrane pore formation.Philos Trans R Soc Lond B Biol Sci. 2017 Aug 5;372(1726):20160219. doi: 10.1098/rstb.2016.0219. Philos Trans R Soc Lond B Biol Sci. 2017. PMID: 28630158 Free PMC article. Review.
-
Coacervation of poly-electrolytes in the presence of lipid bilayers: mutual alteration of structure and morphology.Chem Sci. 2022 Jun 16;13(26):7933-7946. doi: 10.1039/d2sc02013k. eCollection 2022 Jul 6. Chem Sci. 2022. PMID: 35865903 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
