

Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome
- PMID: 20137775
- PMCID: PMC2820190
- DOI: 10.1016/j.ajhg.2010.01.013
Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome
Abstract
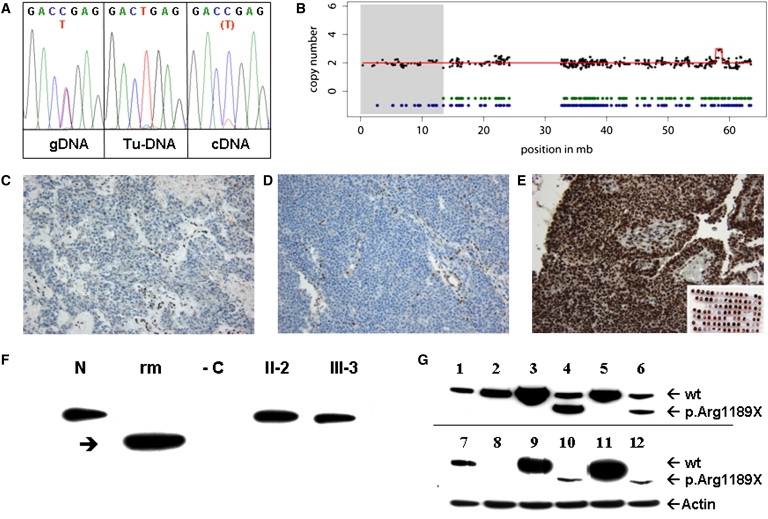
Rhabdoid tumors of early infancy are highly aggressive with consequent poor prognosis. Most cases show inactivation of the SMARCB1 (also known as INI1 and hSNF5) tumor suppressor, a core member of the ATP-dependent SWI/SNF chromatin-remodeling complex. Familial cases, described as rhabdoid tumor predisposition syndrome (RTPS), have been linked to heterozygous SMARCB1 germline mutations. We identified inactivation of another member of the SWI/SNF chromatin-remodeling complex, its ATPase subunit SMARCA4 (also known as BRG1), due to a SMARCA4/BRG1 germline mutation and loss of heterozygosity by uniparental disomy in the tumor cells of two sisters with rhabdoid tumors lacking SMARCB1 mutations. SMARCA4 is thus a second member of the SWI/SNF complex involved in cancer predisposition. Its general involvement in other tumor entities remains to be established.
Copyright (c) 2010 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Jackson E.M., Sievert A.J., Gai X., Hakonarson H., Judkins A.R., Tooke L., Perin J.C., Xie H., Shaikh T.H., Biegel J.A. Genomic analysis using high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin. Cancer Res. 2009;15:1923–1930. - PMC - PubMed
-
- Versteege I., Sévenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
