

Visualization of feline calicivirus replication in real-time with recombinant viruses engineered to express fluorescent reporter proteins
- PMID: 20137802
- PMCID: PMC2855553
- DOI: 10.1016/j.virol.2009.12.035
Visualization of feline calicivirus replication in real-time with recombinant viruses engineered to express fluorescent reporter proteins
Abstract
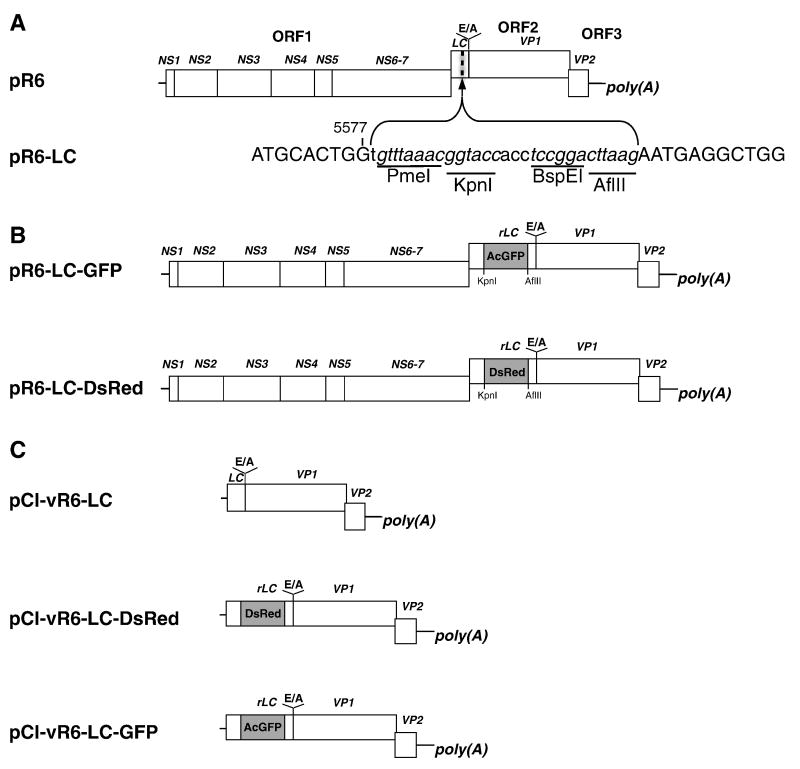
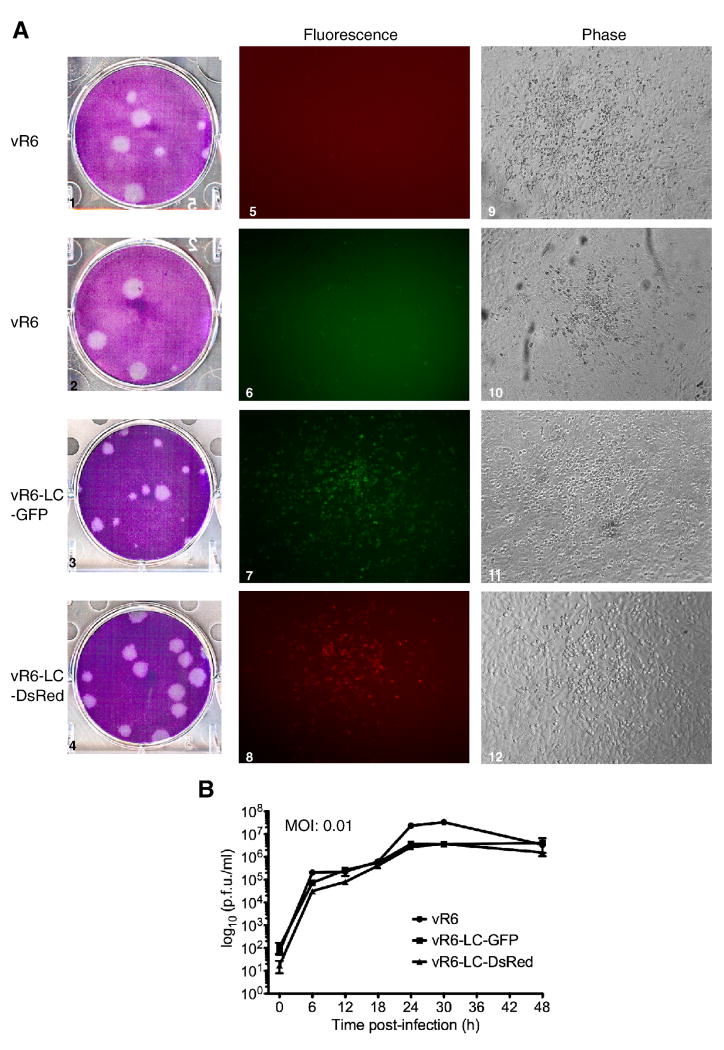
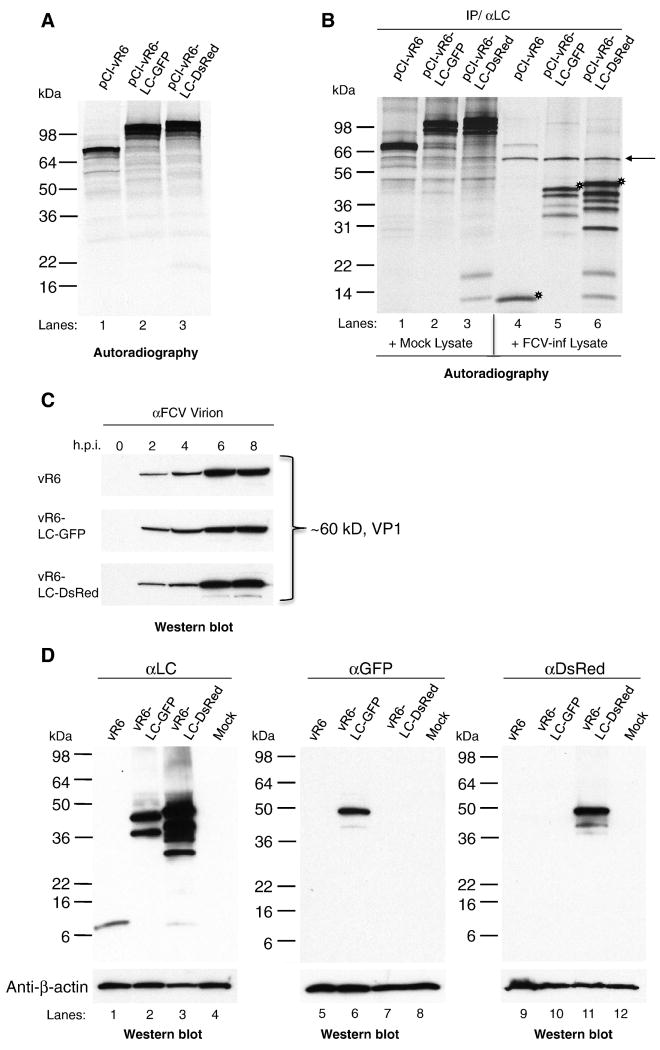
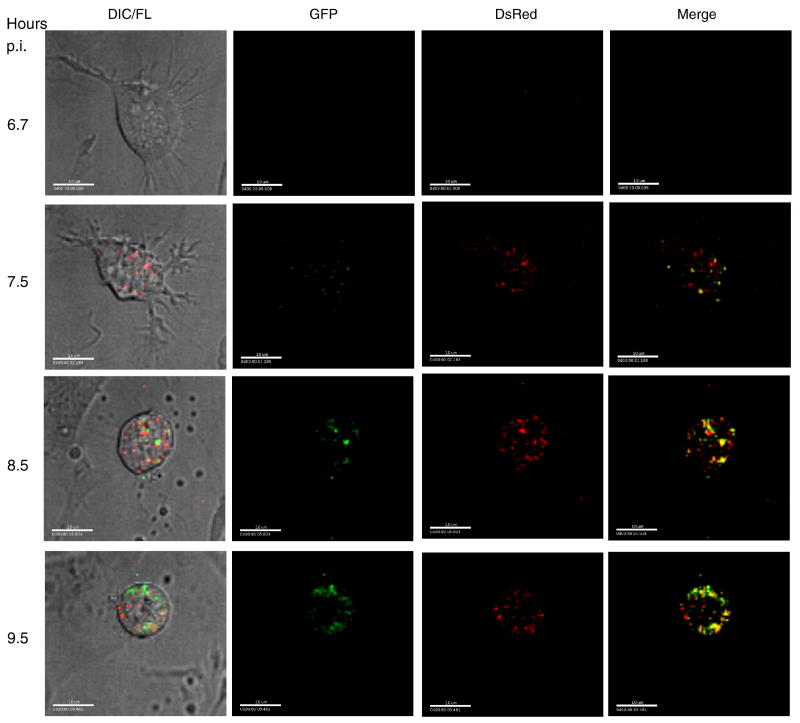
Caliciviruses are non-enveloped, icosahedral viruses with a single-stranded, positive sense RNA genome. Transposon-mediated insertional mutagenesis was used to insert a transprimer sequence into random sites of an infectious full-length cDNA clone of the feline calicivirus (FCV) genome. A site in the LC gene (encoding the capsid leader protein) of the FCV genome was identified that could tolerate foreign insertions, and two viable recombinant FCV variants expressing LC fused either to AcGFP, or DsRedFP were recovered. The effects of the insertions on LC processing, RNA replication, and stability of the viral genome were analyzed, and the progression of a calicivirus single infection and co-infection were captured by real-time imaging fluorescent microscopy. The ability to engineer viable recombinant caliciviruses expressing foreign markers enables new approaches to investigate virus and host cell interactions, as well as studies of viral recombination, one of the driving forces of calicivirus evolution.
Published by Elsevier Inc.
Figures

References
-
- Andino R, Silvera D, Suggett SD, Achacoso PL, Miller CJ, Baltimore D, Feinberg MB. Engineering poliovirus as a vaccine vector for the expression of diverse antigens. Science. 1994;265(5177):1448–51. - PubMed
-
- Biery MC, Lopata M, Craig NL. A minimal system for Tn7 transposition: the transposon-encoded proteins TnsA and TnsB can execute DNA breakage and joining reactions that generate circularized Tn7 species. J Mol Biol. 2000;297(1):25–37. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
